Aller au contenu principal : Appuyer
sur Retour
François JACOB, David PERRIN, Carmen SANCHEZ, Jacques MONOD
II. La coordination de l'expression génétique
![]() Si
je suis devenu généticien c'est que, par mon intérêt d'entomologiste,
j'avais déjà abordé certains aspects qui me paraissaient essentiels
pour expliquer l'évolution et la survie des insectes. Dans mon premier
livre, Ordre et Dynamique du Vivant, fruit des quatre années
du cours d'initiation à la biologie que nous avions créé à l'école
Polytechnique avec Maurice Guéron, j'ai illustré comment le mimétisme
batésien protège une sous-espèce de Papilio dardanus. Ce qui
m'avait frappé à l'époque où je découvrais par moi-même ce phénomène
bien connu des entomologistes, c'est comment une variabilité
préexistante (contingente a priori) pouvait permettre a
posteriori la stabilisation sélective d'une forme localement
adéquate à un environnement donné. Et il me semblait que la génétique
devait être le moyen idéal de comprendre comment se crée la
variabilité et comment opère la sélection.
Si
je suis devenu généticien c'est que, par mon intérêt d'entomologiste,
j'avais déjà abordé certains aspects qui me paraissaient essentiels
pour expliquer l'évolution et la survie des insectes. Dans mon premier
livre, Ordre et Dynamique du Vivant, fruit des quatre années
du cours d'initiation à la biologie que nous avions créé à l'école
Polytechnique avec Maurice Guéron, j'ai illustré comment le mimétisme
batésien protège une sous-espèce de Papilio dardanus. Ce qui
m'avait frappé à l'époque où je découvrais par moi-même ce phénomène
bien connu des entomologistes, c'est comment une variabilité
préexistante (contingente a priori) pouvait permettre a
posteriori la stabilisation sélective d'une forme localement
adéquate à un environnement donné. Et il me semblait que la génétique
devait être le moyen idéal de comprendre comment se crée la
variabilité et comment opère la sélection.
Les découvertes qui ont jalonné les chemins de la génétique moléculaire viennent de la nature analytique de ses approches. On a mis en évidence une collection d'objets pertinents, et on a, mais plus rarement, cherché à les mettre en relation entre eux. J'ai recherché s'il existe des familles de contrôles métaboliques généraux permettant de comprendre comment les divers composants des synthèses macromoléculaires fonctionnent ensemble. Pour cela j'ai choisi le système cellulaire le mieux connu et en un certain sens le plus simple, le colibacille, Escherichia coli. Ma conjecture était que s'il existe un mécanisme autre que la seule compétition entre les divers opérons d'une cellule pour recruter les composants de la machinerie biosynthétique, ce mécanisme doit se manifester via l'organisation hiérarchique d'une cascade de régulations entrelacées. Ce n'est qu'après avoir découvert l'organisation des gènes dans les génomes que j'ai commencé à comprendre comment se faisait ce lien de l'architecture des chromosomes avec l'architecture de la cellule.
Cette conjecture permettait de construire des expériences (en couplant génétique et physiologie) pour en mesurer la validité. Comme je n'avais aucun espoir a priori de tomber d'emblée sur un élément central de ce contrôle, je ne pouvais progresser que par essais et erreurs. Pour commencer, je supposai qu'il n'existait pas de réelle redondance entre des signaux différents, et que si l'on rencontrait cette situation, cela signifiait que la fonction de l'un des signaux redondants était encore inconnue, illustrant une sorte de "ponctuation secondaire" de l'expression génétique (comme les virgules dans un texte écrit), essentielle pour déterminer la signification de l'information véhiculée par le texte. Je choisis pour point de départ l'étude d'un signal métabolique redondant, la formylation de la méthionine portée par l'ARN de transfert du début de la traduction chez E. coli.
L'approche était simple : il s'agissait de trouver des mutants capables de croître dans des conditions où la formylation n'aurait pas lieu. A partir de leur analyse on pourrait comprendre le rôle de ce signal. Mes premières expériences, dans un coin du laboratoire de H. Buc à l'Institut Pasteur, profitant des conseils glanés auprès de mes collègues, ont donc consisté à construire un milieu approprié pour la sélection de ces mutants. A ma grande surprise, j'en isolai plusieurs milliers. Il devenait aisé de tester la validité de la conjecture en cherchant si parmi ces mutants certains étaient altérés dans une étape des synthèses macromoléculaires. Or je trouvai que, si plus de 90% des mutants étaient altérés au locus thyA (thymidylate synthétase) pour des raisons faciles à expliquer au vu de l'organisation générale du métabolisme de l'acide folique et du flux métabolique des groupes à un atome de carbone, près de 5% des mutants résistaient à la rifampicine (et avaient donc une ARN polymérase altérée) et quelques uns résistaient soit à la streptomycine, soit à la spectinomycine (et avaient donc un ribosome altéré). Inversement, je trouvai que sur le milieu sélectif initial (qui ne permettait pas la formylation de l'ARNt démarreur) les souches résistantes à la streptomycine (isolées sur l'antibiotique) ou certaines souches résistantes à la rifampicine pouvaient croître.
Ainsi, au moins pour la phénoménologie, observait-on le couplage des différentes synthèses macromoléculaires via le métabolisme de l'acide folique et des composés à un carbone impliqués dans la formylation de la méthionine portée par l'ARNt démarreur. Pour aller plus avant il fallait disséquer le phénomène en sous-phénomènes plus simples. Pour cela je choisis deux approches, et à l'Institut de Biologie Physico-Chimique où je commençais à fonder un petit groupe avec l'aide de deux étudiants (Marc Uzan et Hans Uffe Petersen), j'entrepris l'étude génétique du couplage traduction / transcription via le métabolisme des dérivés à un carbone (en particulier des acides aminés sérine, méthionine et glycine) et l'étude biochimique du rôle de la formylation au démarrage de la synthèse des protéines.
Toute carence en acide aminé se traduit chez le colibacille par l'arrêt immédiat de la transcription des ARN stables (ARN ribosomiques et ARN de transfert) concomitant de l'arrêt de la traduction (phénotype "stringent"). Puisque l'ARN polymérase était impliquée dans la formylation au démarrage de la traduction, il fallait chercher si les acides aminés impliqués dans le métabolisme de l'acide folique (composés à un-carbone) avaient un comportement particulier vis-à-vis du couplage entre traduction et transcription. C'était facile puisque le gène responsable du couplage strict, relA, était connu. Marc Uzan au cours de son travail de DEA puis au début de sa thèse de troisième cycle, mit ainsi en évidence un phénomène inattendu : les souches relA+ sont transitoirement sensibles au mélange des trois acides aminés (sérine + méthionine + glycine), alors que les souches relA- sont totalement incapables de croître sur ce mélange. Par la suite, M. Uzan démontra que la sensibilité des souches relA- est due à leur incapacité de déréprimer l'opéron ilvGEDA nécessaire à la synthèse de l'isoleucine et de la valine. En parallèle, l'effet inhibiteur des trois acides aminés était levé par l'addition d'isoleucine dans le milieu, un acide aminé qui n'a aucun rapport métabolique commun avec les précédents !
Cette série d'observations inattendues nous a amenés à tenter de comprendre les relations entre la sérine d'une part et l'isoleucine d'autre part et de relier le métabolisme de ces acides aminés à des contrôles plus généraux. Cela m'a conduit, au moyen d'une technique nouvelle d'enrichissement en mutants conditionnels, à isoler des mutants ultra-sensibles à la sérine. Parmi ceux-ci un mutant particulier, hns, devait jouer un rôle particulier au laboratoire. Plutôt que suivre le cours chronologique des expériences, sautons quelques années. Je n'ai finalement compris les raisons de l'effet sérine qu'en janvier 2006, à la suite de nombreux détours, et en particulier grâce à mon activité d'annotation des fonctions génomiques. Cet effet est dû à l'incompatibilité mutuelle de dérivés de la sérine et de co-facteurs du métabolisme, un phénomène semblable au phénomène de frustration identifiés par les physiciens dans d'autres domaines. D'une part la transamination de la sérine conduit à au 3-hydroxypyruvate, inhibiteur des enzymes à thiamine (via la formation d'un dérivé suicide), et d'autre part les réactions utilisant la sérine (synthèse de la cystéine et du tryptophane en particulier) conduisent à la synthèse d'un intermédiaire très réactif, aminoacrylate / iminopropionate, qui peut agir sur un grand nombre de cibles, loin d'être toutes identifiées en 2017.
Au moment de mon installation à l'Institut Pasteur (en 1983) Philippe Lejeune, de l'Université Catholique de Louvain-la-Neuve, m'annonçait qu'il avait découvert un gène conférant une grande sensibilité à la sérine et souhaitait l'étudier. Après bien des expériences, parce que la mutation qu'il avait isolée était une délétion (et pouvait donc mettre en cause plusieurs gènes), nous avons trouvé que le responsable de la sensibilité à la sérine était le gène bglY (alias osmZ, drdX ou pilG, et désormais appelé hns), codant une protéine d'importance majeure. Cette protéine avait été isolée par Michel Jacquet et Régine Cukier-Kahn, dans le laboratoire d'Henri Buc, comme se liant fortement à l'ADN au sein des complexes de transcription. Elle était considérée comme une protéine importante pour le contrôle de la transcription (et de la virulence de nombreuses entérobactéries). Appelée initialement H1, parce qu'elle faisait partie de la fraction résistante à la température des complexes de transcription —cela a rarement (peut-être jamais) été souligné— c'est en raison de cette homonymie accidentelle et de son aptitude à se lier à l'ADN qu'elle a été longtemps considérée comme une protéine "de type histone". Elle est aujourd'hui appelée H-NS. Le gène hns contrôle l'expression de gènes responsables de la sensibilité à la sérine. La protéine H-NS sonde l'état de l'environnement (variation de la température, de la richesse du milieu, de la présence d'oxygène, de l'osmolarité, de l'acidité...). Elle agit au travers d'une interaction fonctionnelle avec une protéine à effet très pléiotrope, LRP (codée par le gène lrp), qui contrôle l'expression de gènes pde dégradation de la sérine, mais aussi celle d'une acétohydroxyacide synthase spécifique de la synthèse de l'isoleucine (travail de Mark Levinthal, en année sabbatique au laboratoire en 1992, et travaux poursuivis en collaboration avec son laboratoire à l'Université Purdue, où j'ai effectué plusieurs séjours). Par ailleurs, l'expression de près de 200 gènes est spécifiquement affectée par la mutation hns (Philippe Bertin, P. Lejeune). Nous avons enfin fait la découverte —toujours inexpliquée en 2019 et peut-être indirecte— qu'il intervient dans la maintenance de l'intégrité de l'ADN, puisque son absence augmente d'un facteur 100 la fréquence des délétions spontanées, (ce qui complique singulièrement l'étude génétique des mutants de ce gène) !
La difficulté de l'étude de hns nous a amenés à nous concentrer sur des cibles de l'effet sérine situées plus en aval, d'une part, et d'autre part à privilégier l'analyse d'opérons spécifiques sur lesquels l'action de hns se fait sentir. Cela nous a permis de découvrir qu'outre son effet de contrôle négatif, la protéine H-NS exerce un contrôle positif dans un certain nombre de cas (travaux de P. Bertin). H-NS contrôle ainsi la genèse des flagelles (et donc la chimiotaxie), mais aussi la croissance en l'absence d'oxygène. Cette dernière découverte se relie à la véritable fonction de cette protéine, intervenant particulièrement dans la résistance à l'acidité. Le développement de la transcriptomique et de la protéomique nous a permis de mieux caractériser les effets de H-NS, de comprendre comment se fait le lien avec ces systèmes. L'action directe de H-NS au démarrage de la transcription de certains gènes n'est qu'un effet de recrutement d'une activité complémentaire à un effet beaucoup plus général impliquant des complexes comprenant des ARN messagers spécifiques ou des petits ARN non traduits. H-NS est impliquée dans le contrôle de la disponibilité en protons du périplasme de beaucoup de bactéries à coloration de Gram négative. Nos travaux ultérieurs ont été consacrés à cette exploration, en particulier via l'étude génomique d'une bactérie psychrophile isolée dans l'Antarctique, Pseudoalteromonas haloplanktis.
Revenons à l'effet sérine, et au rôle de la formylation. Dans le cas de l'effet de mutants du ribosome sur la formylation, nous pouvions éprouver nos hypothèses à partir d'expériences biochimiques. Avec Uffe Petersen, nous avons analysé in vitro le rôle de la formylation dans la synthèse du premier lien peptidique. L'idée était de différencier les ribosomes sauvages des ribosomes résistants à la streptomycine, puisque la mutation correspondante permettait la croissance dans un milieu où la formylation n'a pas lieu. Avec les ribosomes sauvages nous avons identifié deux mécanismes distincts. Le premier met en jeu les sous-unités ribosomiques dissociées avec l'ordre des événements suivant : la sous-unité 30S attache l'ARN messager et l'ARNt démarreur, puis la sous-unité 50S se lie et, en présence d'un ARNt chargé (ou de puromycine) on observe la formation du premier lien peptidique. Ce mécanisme semble très peu sensible à la formylation. Un deuxième mécanisme met en jeu les ribosomes préassociés 70S. Cette fois le démarrage de la traduction ne peut se faire qu'en présence de l'ARNt démarreur formylé.
Ces observations était paradoxales puisqu'on pensait que chez les bactéries la synthèse des protéines commence avec des ribosomes dissociés et qu'elle requiert la présence d'un groupe formyl- sur la méthionine initiale. L'étude biochimique du démarrage de la traduction sur les ribosomes 70S nous a conduits à découvrir un équilibre conformationnel entre deux formes, l'une majoritaire et inactive pour le démarrage de la traduction et l'autre, minoritaire et active. Le Met-ARNt démarreur non formylé serait capable de se lier également sur les deux formes, alors que la formylation l'empêcherait de se lier sur la forme inactive. Le groupe formyl- apparaît alors comme un effecteur allostérique positif déplaçant l'équilibre vers la forme qui déclenche le départ de la traduction. Ces expériences conduisent à imaginer un rôle spécifique de la formylation, autre que la simple reconnaissance du codon du début de traduction. Au cours de la traduction des messagers polycistroniques, il existe deux mécanismes distincts de démarrage, le départ de la synthèse polypeptidique sur le premier cistron se ferait à l'aide de ribosomes prédissociés et dépendrait peu de la formylation ; au contraire, pour les cistrons suivants, le départ de la traduction utiliserait, le plus souvent, des ribosomes associés, 70S, et la formylation serait alors essentielle. Citant ce travail pionnier, la démonstration de cet effet allostérique sur le démarrage de la traduction à partir de ribosomes 70S a été confirmé quarante ans plus tard par le laboratoire de Knud Nierhaus.
On pouvait mettre à l'épreuve cette conjecture. L'opéron lactose est constitué de trois cistrons et le produit de deux d'entre eux peut être dosé avec une grande précision. Avec Agnès Ullmann (1927-2019) nous avons donc dosé la β-galactosidase et surtout la méthylthiogalactoside transacétylase avec sa technique de dosage unique au monde. Il suffisait d'ajouter à la culture bactérienne induite pour l'expression de l'opéron lactose des quantités croissantes d'un inhibiteur de la formylation puis de doser la β-galactosidase et l'acétylase. Si l'hypothèse de départ était fondée on attendait que le rapport β-galactosidase / transacétylase augmentât. Dès la première conversation que j'eus avec elle, A. Ullmann m'apprit qu'elle avait déjà fait cette expérience dix ans auparavant, et que le résultat (non publié) était celui que j'attendais ! Elle me proposa alors que nous collaborions en étudiant plusieurs moyens de faire varier la formylation et, bien sûr, en revenant au comportement de la mutation qui avait motivé les expériences initiales, à savoir la résistance à la streptomycine. C'est ainsi que commença une collaboration qui s'est développée en parallèle avec le développement de nos laboratoires et qui a duré plus de quinze ans.
Pour mettre en évidence le phénomène de polarité métabolique, nous avons utilisé une inhibiteur de la formylation, le trimethoprim, puis je construisis un certain nombre de souches bactériennes altérées dans l'utilisation des substrats ou précurseurs de la formylation. Nous avons observé une forte polarité (décroissance de l'expression de la transacétylase par rapport à la β-galactosidase) à chaque fois que le taux intracellulaire de formyl-tetrahydrofolate (le donneur de formyl-) décroissait. De plus nous avons trouvé que les mutants résistants à la streptomycine échappaient à ce phénomène (ce qui démontre une implication de la traduction) et que cela est dû au fait que les ribosomes de souches résistantes à cet antibiotique sont naturellement dissociés ( des sous-unités libres dans la cellule permettent la reconnaissance et le démarrage de la traduction au début de n'importe quel cistron).
Ces expériences expliquent l'organisation en opérons polycistroniques
chez les procaryotes. On peut alors remarquer deux classes de régions
intercistroniques. Ou bien cette région est assez longue et souvent
possède une ou plusieurs structures de type tige et boucle, ou bien
cette région est très courte et parfois même formées de cistrons qui
se recouvrent. Dans le premier cas le démarrage de la traduction des
deux cistrons considérés se fait de façon indépendante, alors que dans
le deuxième cas le ribosome qui termine la traduction du premier
cistron commence celle du suivant sans se dissocier, à condition que
le taux de formylation de l'ARNt démarreur soit suffisant. Après
l'avènement de la génomique ces travaux sont revenus sur le devant de
la scène : l'organisation de l'opéron pyrH frr, qui code
l'uridylate kinase et le facteur de recyclage du ribosome, suggère que
les ribosomes![]() 70S ne se dissocient pas au cours de la lecture d'un opéron
polycistronique, mais que c'est au moment de la terminaison de la
transcription, lorsqu'une tige et boucle suivie d'un poly U indique la
fin du messager, que le recyclage a lieu. Nous avons conjecturé que
l'UTP est un inhibiteur de ce facteur. Ce travail, entrepris à partir
de la base de données spécialisée que nous avons construite pour
rassembler tout ce qui est connu sur la séquence du chromosome d'E.
coli, Colibri, nous conduisit (nous le verrons
plus loin), à penser qu'il existe un lien fort entre l'ordre des gènes
dans le chromosome, et l'architecture de la cellule.
70S ne se dissocient pas au cours de la lecture d'un opéron
polycistronique, mais que c'est au moment de la terminaison de la
transcription, lorsqu'une tige et boucle suivie d'un poly U indique la
fin du messager, que le recyclage a lieu. Nous avons conjecturé que
l'UTP est un inhibiteur de ce facteur. Ce travail, entrepris à partir
de la base de données spécialisée que nous avons construite pour
rassembler tout ce qui est connu sur la séquence du chromosome d'E.
coli, Colibri, nous conduisit (nous le verrons
plus loin), à penser qu'il existe un lien fort entre l'ordre des gènes
dans le chromosome, et l'architecture de la cellule.
Ces phénomènes ravivaient un intérêt déjà ancien pour l'AMP cyclique et son récepteur. Comprendre le rôle des médiateurs de ce type peut se faire via l'étude de leurs cibles, mais il était plus efficace, plutôt que de faire un catalogue des cibles, de s'attacher à comprendre la régulation de leur synthèse. L'adénylcyclase devenait par ailleurs une enzyme d'intérêt médical puisque plusieurs études l'ont impliquée comme élément majeur de la virulence bactérienne. J'ai donc choisi d'en entreprendre l'étude génétique, en collaboration avec A. Ullmann et O. Bârzu qui ont pris en charge les aspects plus particulièrement liés à la biochimie de l'enzyme.
Nous avons cloné et analysé le gène cyaA codant la sous-unité
catalytique de la cyclase (thèse de 3ème cycle, Anne Roy). En
collaboration avec Hiroji Aiba à Kyoto nous avons ensuite déterminé la
séquence complète du gène (ce qui à l'époque était encore difficile).
![]() Les
gènes cyaA et dapF sont transcrits dans le même sens,
les autres dans le sens opposé. Par ailleurs le gène cyaA est
bien transcrit mais mal traduit, à partir d'un codon de démarrage
inhabituel, UUG. Durant la phase exponentielle, malgré une
transcription efficace, la quantité d'enzyme synthétisée est très
faible, elle augmente brusquement au moment de l'entrée en phase
stationnaire. C'est la traduction qui en limite l'expression, et une
région du début de la zone traduite est impliquée dans ce contrôle.
Nous avons montré une grande similitude dans l'organisation du gène et
de sa région de contrôle et une conservation quasi parfaite de la
séquence de nucléotides recouvrant le codon de démarrage de la
traduction UUG chez d'autres entérobactéries : Erwinia
chrysanthemi, Yersinia intermedia et Yersinia pestis.
Cela nous a permis de cloner et de séquencer le gène chez Proteus
mirabilis après criblage par hybridation de l'ADN. L'isolement
du gène de la cyclase de Pasteurella multocida (en
collaboration avec M. Mock) nous a montré que si la protéine était
bien apparentée aux cyclases des entérobactéries (même organisation du
gène en deux domaines, et 30% d'identité de séquence), les régions de
contrôle, en amont et en aval du gène, étaient tout-à-fait
différentes. Nous avons déterminé la séquence de la région homologue
chez les trois entérobactéries, et nous avons découvert en aval du
gène cyaA un gène, cyaY, transcrit en sens opposé. En
collaboration avec Mark Borodovsky de Georgia Tech, nous avons
démontré la réalité de cyaY. Ce gène code une protéine
apparentée au gène responsable d'une maladie humaine, l'ataxie de
Friedreich. Nous savons aujourd'hui que le produit de ce gène
intervient dans le métabolisme des noyaux fer-soufre.
Les
gènes cyaA et dapF sont transcrits dans le même sens,
les autres dans le sens opposé. Par ailleurs le gène cyaA est
bien transcrit mais mal traduit, à partir d'un codon de démarrage
inhabituel, UUG. Durant la phase exponentielle, malgré une
transcription efficace, la quantité d'enzyme synthétisée est très
faible, elle augmente brusquement au moment de l'entrée en phase
stationnaire. C'est la traduction qui en limite l'expression, et une
région du début de la zone traduite est impliquée dans ce contrôle.
Nous avons montré une grande similitude dans l'organisation du gène et
de sa région de contrôle et une conservation quasi parfaite de la
séquence de nucléotides recouvrant le codon de démarrage de la
traduction UUG chez d'autres entérobactéries : Erwinia
chrysanthemi, Yersinia intermedia et Yersinia pestis.
Cela nous a permis de cloner et de séquencer le gène chez Proteus
mirabilis après criblage par hybridation de l'ADN. L'isolement
du gène de la cyclase de Pasteurella multocida (en
collaboration avec M. Mock) nous a montré que si la protéine était
bien apparentée aux cyclases des entérobactéries (même organisation du
gène en deux domaines, et 30% d'identité de séquence), les régions de
contrôle, en amont et en aval du gène, étaient tout-à-fait
différentes. Nous avons déterminé la séquence de la région homologue
chez les trois entérobactéries, et nous avons découvert en aval du
gène cyaA un gène, cyaY, transcrit en sens opposé. En
collaboration avec Mark Borodovsky de Georgia Tech, nous avons
démontré la réalité de cyaY. Ce gène code une protéine
apparentée au gène responsable d'une maladie humaine, l'ataxie de
Friedreich. Nous savons aujourd'hui que le produit de ce gène
intervient dans le métabolisme des noyaux fer-soufre.
Avec la construction d'un gène hybride entre le gène cyaA et le gène lacZ nous avons montré que la protéine hybride purifiée synthétise l'AMP cyclique et hydrolyse le lactose : nous venions d'identifier pour la première fois le gène d'une adénylcyclase. Grâce à ces constructions, nous avons étudié le comportement de gènes tronqués de la cyclase et nous avons trouvé qu'on peut tronquer plus de 50% de la partie 3' terminale du gène sans perdre l'activité cyclase. On observe cependant que la régulation de l'activité par le glucose est abolie dès que quelques pour cents du gène sont détruits, alors que l'activité de l'enzyme augmente. La protéine chez E. coli est formée de deux domaines, le domaine amino-terminal est doué d'activité cyclase et le domaine carboxy-terminal est le relais de l'effet inhibiteur du glucose sur la synthèse d'AMP cyclique. Nous avons défini ensuite les limites du domaine catalytique actif, montrant que la régulation est une inhibition tonique établie par le domaine carboxy-terminal. L'un des aspects les plus remarquables est la relation entre l'activité de l'adénylcyclase et le transport des sucres apparentés au glucose. L'objet d'une partie de nos études a donc été de comprendre dans le détail l'organisation fonctionnelle du système de transport de ces sucres et ses relations avec le métabolisme.
Afin d'élargir notre étude phylogénétique nous avons étudié l'adénylcyclase toxique de Bordetella pertussis, l'agent de la coqueluche. Après de nombreuses tentatives infructueuses d'isolement dans une souche de colibacille défective en adénylcyclase, nous avons mis à profit le fait que l'enzyme de B. pertussis est activée par une protéine eucaryote, la calmoduline. J'ai construit une souche de colibacille défective en cyclase, et portant un plasmide codant une calmoduline synthétique. Philippe Glaser a alors criblé dans cette souche une banque de B. pertussis créée dans un plasmide compatible, ce qui a permis l'isolement du gène. Cette expérience ancêtre du "double-hybride" utilisant une complémentation originale à trois partenaires, a aussitôt été répétée, en collaboration avec M. Mock, avec l'ADN d'un autre pathogène, Bacillus anthracis, l'agent du charbon. J'ai par ailleurs fait l'expérience symétrique pour isoler le messager de la calmoduline humaine.
La séquence des gènes correspondants a montré qu'ils sont très éloignés, mais apparentés l'un à l'autre, et totalement distincts des cyclases des entérobactéries. Chez B. anthracis, la protéine (800 acides aminés, comprenant un peptide signal) a trois domaines, le domaine catalytique étant central, alors que chez B. pertussis, la protéine, très grande (1706 acides aminés) porte l'activité catalytique dans les 400 résidus amino-terminaux. Cette organisation est liée au mode de sécrétion de ces toxines, classique, au moyen d'un peptide signal, dans le premier cas, original et nécessitant la présence de trois produits de gènes supplémentaires dans le second. Le mécanisme de la sécrétion de B. pertussis est semblable à celui de toxines comme l'hémolysine de E. coli et met en jeu un dispositif complexe permettant le passage au travers des deux membranes de ces organismes à Gram négatif (sécrétion de type I). Ces succès nous ont conduit, à nous interroger sur l'usage militaire de cette toxine, dans un contexte qui n'y a alors prêté aucune attention.
Notre étude de la cyclase de B. pertussis a été développée très en détail, en vue de la fabrication d'un composant nouveau d'un vaccin contre la coqueluche, puis pour comprendre le mécanisme de la cyclisation de l'ATP en AMP cyclique, et de l'activation par la calmoduline (en collaboration avec Octavian Bârzu). Nous avons identifié trois régions essentielles pour l'activité ou pour l'activation de l'enzyme et caractérisé son mode d'interaction avec la calmoduline. Nous avons construit plusieurs dizaines de mutants pour mettre en évidence les résidus et les structures secondaires les plus importantes, et nous en arrivons à un modèle où la calmoduline rapprocherait deux domaines de la protéine enserrant le site catalytique, lui-même apparenté à celui des phosphofructokinases. Cette hypothèse a été confortée par l'observation que l'activité de la protéine pouvait être reconstituée par l'association non covalente de deux fragments, en eux-mêmes non fonctionnels exactement comme cela avait été montré par A. Ullmann avec la beta-galactosidase (complémentation alpha-oméga).
Nous nous trouvions donc en présence de deux classes d'adénylcyclases. Il était s tentant d'en étudier la parenté avec les enzymes homologues des eucaryotes, d'autant qu'il semblait exister une parenté immunochimique entre les enzymes de B. pertussis et une enzyme cérébrale. Nous avons alors cloné et séquencé le domaine catalytique du gène de l'adénylcyclase de la levure Saccharomyces cerevisiae (travail de Patrick Masson). Notre étude a montré que le domaine catalytique était carboxy-terminal. Curieusement, cette adénylcyclase ne ressemblait à aucune des deux classes que nous avions déjà identifiées. En parallèle, nous avons étudié deux autres cyclases, elles aussi clonées par complémentation d'un mutant défectif de E. coli, celle de Rhizobium meliloti (thèse d'Annie Beuve, en collaboration avec Fergal O'Gara de l'Université de Cork en Irlande) et celle de Brevibacterium liquifaciens (en collaboration avec Elizabeth Peters et Tom Blundell, du Birkbeck College à Londres) et nous avons découvert qu'elles étaient apparentées à la cyclase de levure. Une deuxième cyclase de R. meliloti a plus tard été isolée et séquencée, en collaboration avec F. O'Gara. En parallèle, en collaboration avec Bernard Lubochinsky nous avons identifié chez la bactérie Stigmatella aurantiaca (myxobactérie différenciée) deux adénylcyclases très éloignées l'une de l'autre. Ensuite, en collaboration avec A. Ullmann et Charles Thompson, nous avons isolé le gène de l'enzyme de Streptomyces coelicolor, recherché de puis de nombreuses années par bien des laboratoires dans le monde. Nous avons montré qu'il n'y avait qu'un seul gène cya fonctionnel chez cette bactérie, et que l'AMP cyclique était impliqué dans la différenciation productrice du mycelium aérien. Un peu auparavant, plusieurs laboratoires décrivaient des adényl- et guanyl-cyclases mammifères, qui faisaient partie de cette même classe. Nous avons ainsi découvert trois classes d'adénylcyclases, l'une d'entre elles étant commune aux eucaryotes et aux bactéries Gram+ et Gram-. Cette dernière observation montre que l'AMP cyclique a joué un rôle très tôt au cours de l'évolution, mais sans doute pas seulement comme régulateur de l'expression des gènes. Afin de tester l'idée d'une origine commune, nous avons cherché à faire évoluer l'adénylcyclase de Rhizobium vers une activité guanylcyclase. Nous avons mis au point un crible génétique dans lequel les souches réceptrices de plasmides mutagénisés porteurs du gène de l'adénylcyclase ne peuvent croître que si elles synthétisent le GMP cyclique. Nous avons trouvé des enzymes mutées de ce type, puis réalisé un aller et retour entre les activités adénylcyclase et guanylcyclases de variants d'une même protéine (thèse de A. Beuve).
Plus tard Christian Vivarès a isolé deux gènes d'adénylcyclase chez Aeromonas hydrophila. L'un d'entre eux fait partie de la classe des protéines des entérobactéries et se trouve entre les enzymes des entérobactéries et celle de P. multocida. L'autre est tout à fait énigmatique, et correspond à une classe d'adénylcyclases entièrement nouvelle, semblable au produit de gènes d'archébactéries. Il s'agit là d'un cas de convergence évolutive remarquable, dont nous ne comprenons pas encore la fonction : les protéines apparentées chez c'autres organismes sont incapables synthètiser l'AMP cyclique. Cette cyclase est une petite protéine, dont l'activité optimale se déroule à 65°C (alors qu'A. hydrophila ne croît pas à plus de 30°C), et à pH alcalin. L'inactivation du gène correspondant, même dans un contexte totalement dépourvu d'activité adénylcyclase n'a pas permis de découvrir la fonction correspondante. Le gène homologue existe aussi chez Yersinia pestis, qui possède trois gènes d'adénylcyclases, d'origine évolutive différente ! Il s'agit d'une protéine apparentée à la thiamine triphosphate phosphatase, enzyme caractérisée par son action dans le système nerveux central des vertébrés. Cela confirme le fait que connaître la structure, et même l'activité d'une enzyme, ne permet pas d'en connaître la fonction...
Pour approfondir nos réflexions sur la structure des protéines liant les nucléotides et leurs relations avec l'activité enzymatique et la phylogénèse nous avons, en collaboration avec P. Glaser et O. Bârzu, développé le clonage de gènes reconnaissant spécifiquement l'ATP, adénylate kinases, et ensuite, UMP et CMP kinases, afin d'en comprendre le détail du site catalytique et de le comparer à celui des cyclases. Toutes ces approches sont essentielles au développement de la deuxième approche de la coordination de l'expression génétique menée au laboratoire, l'exploration d'un génome bactérien entier.
La synthèse de l'AMP cyclique dépend de la nature de la source de carbone. L'occupation du milieu par les micro-organismes se fait grâce à une spécialisation très poussée qui permet à chacun de faire le tri des métabolites laissés ou produits par les autres, dans des conditions aussi spécifiques que possibles, afin d'éviter les risques d'une compétition directe. La gestion des sources de carbone est cruciale pour l'élaboration de la biomasse et de l'énergie. Et on sait depuis un siècle que certaines sources de carbone, comme le glucose, sont utilisées de préférence au cours du temps, avant les autres, par de nombreux micro-organismes.
Les effets du glucose, en particulier chez le colibacille, ont été groupés en trois classes phénoménologiques. D'abord le glucose exclut les substrats autres que lui-même par un mécanisme indirect, mais très efficace. C'est l'exclusion des inducteurs. Ensuite, ajouté dans une culture croissant sur une autre source de carbone il produit de façon transitoire un arrêt quasi-total de l'expression de nombreux gènes du catabolisme. C'est la répression transitoire. Enfin lorsqu'on cherche à exprimer, avec des inducteurs gratuits (ou dans des souches constitutives), un opéron catabolique sensible au glucose, on ne retrouve qu'une induction partielle. C'est la répression permanente ou répression catabolique proprement dite. Beaucoup de laboratoires ont cherché à identifier les bases moléculaires de ces effets. Le laboratoire de Jacques Monod, étudiant une unité de transcription sensible aux effets du glucose, l'opéron lactose, s'est attaché à comprendre le mécanisme de la répression catabolique. Agnès Ullmann y a en 1968 démontré que l'AMP cyclique, alors récemment découvert, était un médiateur de ce phénomène. Cette découverte reproduite peu après ailleurs dans le monde a été à la base d'un schéma qui s'est rapidement généralisé, où l'AMP cyclique est le médiateur de la répression catabolique. Des travaux issus de ce même laboratoire devaient démontrer cependant que les choses ne pouvaient être aussi simples.
De nombreuses expériences nous avaient indiqué que les protéines impliquées dans le transport du glucose étaient mises en cause dans les divers effets du glucose. C'est l'isolement fortuit d'un mutant défectif dans la synthèse d'AMP cyclique par Rémy Bitoun, qui décida de notre orientation future. En effet ce mutant était très affecté dans le transport du glucose, et, indirectement dans la synthèse d'AMP cyclique. L'étude de la complémentation au moyen d'une banque génomique nous a permis d'isoler l'ensemble des gènes codant la synthèse des composants centraux du système général de transport du glucose. Depuis plus de cinquante ans le système de transport du glucose avait été décrit en termes biochimiques. Le glucose est transporté vectoriellement à l'intérieur de la cellule grâce à sa modification chimique sous forme de glucose-6-phosphate. Cette phosphorylation résulte d'une cascade de phosphorylations de protéines. Le donneur de phosphate initial est le phosphoenolpyruvate (PEP). Une première protéine, l'enzyme I, est phosphorylée; elle phosphoryle à son tour une protéine, HPr, qui phosphoryle directement l'enzyme membranaire qui transporte la source de carbone en la phosphorylant (mannitol, sorbitol, mannose, N-acétyl-glucosamine, etc.), ou transfère son phosphate à une autre protéine, l'enzyme IIA glucose (IIAGlc) qui phosphoryle alors le glucose. Cette cascade complexe appelée PTS est à la base de l'exclusion des inducteurs par le glucose, ou la diminution de l'expression des opérons cataboliques, via le contrôle de la synthèse de l'AMP cyclique.
Les composants membranaires du PTS sont spécifiques de chaque sucre. Au contraire, la fraction cytoplasmique responsable des contrôles pléiotropes est partagée par tous les transports (sauf celui du fructose). Nous avons isolé les gènes ptsH (codant HPr), ptsI (codant l'Enzyme I) et crr (codant l'enzyme IIAGlc). Nous avons montré que, contrairement à l'opinion commune, le gène crr fait partie de la même unité de transcription que les gènes ptsH et ptsI, ce qui implique un contrôle coordonné de leur expression. Hilde De Reuse, au cours de sa thèse de 3ème cycle, a caractérisé un cosmide porteur de la région ptsHI-crr et étudié l'expression de l'opéron ptsHI. La séquence des nucléotides depuis la région de contrôle jusqu'au gène crr a relié ce qui est connu biochimiquement de ces protéines aux gènes correspondants et à la régulation de leur expression. Ensuite l'étude de l'opéron entier (séquence, analyse des transcrits, analyse de la région promotrice par mutagenèse localisée) nous a montré un mode d'expression dans lequel se mêlent les effets du complexe cAMP-CAP et ceux de l'enzyme IIGlc, protéine membranaire responsable de l'entrée du glucose dans la cellule (thèse de Hilde De Reuse). Ainsi, la membrane joue un rôle intégrateur à un double titre, à la fois par le contrôle du niveau d'expression des gènes de la cascade, et par le phénomène de transport. Cela est dû au rôle spécial de l'enzyme IIAGlc qui contrôle l'exclusion des inducteurs et la synthèse du médiateur de la transcription qu'est l'AMPc. L'approche choisie pour comprendre ce rôle a été le criblage de mutations dans le gène crr, d'une part, et, d'autre part, l'analyse de la complémentation de mutants défectifs par des gènes provenant d'organismes hétérologues, de plus en plus éloignés du point de vue phylogénétique. Cela permet de distinguer les effets d'interaction directs (protéines-protéines) et d'effets indirects (médiés par l'expression de protéines intermédiaires) (thèse de Guoqing Zeng).
Afin d'évaluer la généralité de ces résultats, nous avons caractérisé la physiologie et cloné les gènes homologues chez une bactérie phytopathogène très importante pour l'industrie, Xanthomonas campestris. Nos résultats montrent qu'il s'agit d'un système spécifique du fructose tout à fait distinct de celui des entérobactéries, manifestant aussi un effet pléiotrope (mais différent, et ne passant pas par l'AMP cyclique) (thèse de Valérie de Crécy). Des travaux de la société Kelco aux Etats-Unis ont confirmé nos hypothèses concernant l'incorporation du fructose dans ces bactéries (et en particulier l'existence d'une fructose-mannose isomérase).
Pour finir, il nous a semblé essentiel de comprendre la façon dont l'enzyme IIAGlc joue à la fois sur l'exclusion des inducteurs, et sur la synthèse de l'AMP cyclique. Dans ce dernier cas, nous avons démontré (thèses de Sophie Lévy et G. Zeng), grâce au transfert sur le chromosome de délétions construites in vitro, que la synthèse d'AMP cyclique nécessitait non seulement la présence de l'enzyme IIA, mais celle de HPr ou de l'enzyme I. Nous avons recherché des mutants de l'enzyme IIAGlc obtenus soit spontanément soit par mutagenèse in vitro et découvert que l'enzyme IIAGlc agit vraisemblablement via le contrôle du repliement des protéines concernées et non sur leur activité. Il s'agit là d'un nouveau mode de contrôle de l'expression génétique, à l'œuvre dans un grand nombre de systèmes pléiotropes (thèse de GQ Zeng), encore très peu exploré. Par ailleurs nous avons établi que tous les effets connus de modulation de cette activité passaient par la forme phosphorylée de l'enzyme IIAGlc: un résidu aspartate situé à la jonction entre le domaine catalytique et le domaine régulateur est directement impliqué dans la régulation, peut-être au travers d'une phosphorylation.
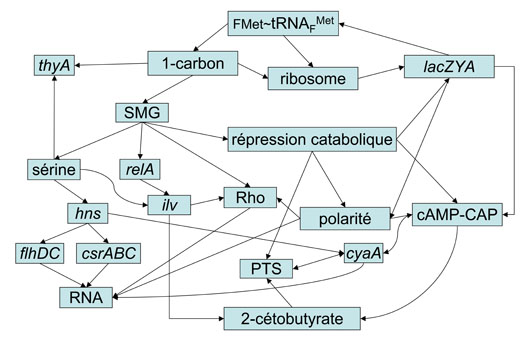
Ma motivation primordiale pour l'étude génétique du colibacille était de chercher des contrôles métaboliques pléiotropes de l'expression génétique. L'AMP cyclique illustre un contrôle de ce type, mais j'espérais mettre en évidence des contrôles en amont qui permettraient de comprendre l'intégration des divers modes de l'expression génétique donnant à la cellule un comportement global. L'étude du cycle des métabolites à un carbone médié par les dérivés de l'acide folique nous avait indiqué une relation inattendue entre la serine et les acides aminés branchés (isoleucine/leucine/valine). Et l'analyse de mutants très sensibles à la sérine m'avait suggéré que l'ATP synthétase de la phosphorylation oxydative pourrait être impliquée dans ce phénomène. Jacques Daniel venait de terminer une étude génétique de mutants de ce système et je fus amené à discuter avec lui des questions qui me préoccupaient et il décida de faire avec moi sa thèse de doctorat d'état sur ce sujet.
Nous n'avions alors que des données phénoménologiques et il semblait difficile de découvrir le lien qui existe entre serine et isoleucine. Après un long travail d'identification nous avons montré qu'un céto-acide précurseur de l'isoleucine, le 2-cétobutyrate est excrété en quantité importante dans une souche crp* (dont la protéine CAP est un activateur des opérons cataboliques en l'absence d'AMP cyclique). Parallèlement je mis au point un milieu de culture qui exaltait la sensibilité à la sérine, et cela nous permit de confirmer que le phénomène étudié impliquait l'AMP cyclique et les différentes sources de carbone. Mieux, j'isolai un mutant qui avait, comme je l'attendais d'un contrôle général, un comportement extrêmement pléiotrope. Philippe Glaser a montré au cours de son travail de DEA qu'il s'agit d'un mutant du facteur Rho, responsable de la terminaison précoce de la transcription (non publié).
Cet ensemble de résultats nous conduisit à étudier plus en détails le rôle du 2-cétobutyrate dans les cellules, à l'aide de cette molécule elle-même, et de divers analogues. Nous avons d'abord pensé qu'il s'agissait d'un effecteur de la répression catabolique, car l'addition de 2-cétobutyrate inhibe très fortement l'expression des opérons cataboliques, mais une analyse plus fine nous montra que le système de transport du glucose (le PTS) est impliqué dans l'inhibition. Après avoir étudié l'action du 2-cétobutyrate et de ses analogues dans de nombreuses conditions physiologiques, et sur de nombreux mutants, et avoir mesuré l'évolution des réserves de molécules phosphorylées en présence du cétoacide nous sommes arrivés au modèle suivant. Le 2-cétobutyrate en excès bloque le système des phosphotransférases et diminue brutalement la concentration intracellulaire d'acétyl-coenzyme A. en inhibant la pyruvate déshydrogénase. Il s'ensuit une cascade d'effets remarquables :
- le transport des sources de carbone est arrêté ;
- le niveau intracellulaire de fructose 1-6 bisphosphate décroît, ce qui entraîne une carence immédiate en aspartate, avec arrêt de la traduction, synthèse de ppGpp, arrêt de la transcription des ARN stables ;
- l'activité de l'adénylcyclase décroît fortement, ce qui empêche toute expression des opérons cataboliques et augmente la polarité de l'expression des opérons polycistroniques.
Ces effets contrôlent l'ensemble du métabolisme cellulaire et de l'expression génétique. Nous nous sommes alors interrogés sur la signification biologique de cet effet, après avoir remarqué que la construction des chemins métaboliques au voisinage du cétobutyrate est particulière :
thréonine acétohydroxyacide
désaminase synthases
thréonine => 2-cétobutyrate => => => isoleucine
+ pyruvate activé
(pyruvate déshydrogénase)
En effet la première enzyme du chemin, la threonine désaminase est retroinhibée par le produit final de la voie métabolique, l'isoleucine. Tout ralentissement en aval du 2-cétobutyrate se traduit immédiatement par une élévation brutale de sa concentration. Or les acétohydroxyacide synthases en aval ont leur activité directement couplée à la pyruvate déshydrogénase, et un excès de 2-cétobutyrate fait précisément décroître l'efficacité de cette voie (inhibition de la synthèse d'acétyl CoA) ! La construction de ce métabolisme est ainsi quasiment explosive et la moindre altération du couplage entre la glycolyse et la respiration devrait mener à une augmentation brutale de la concentration intracellulaire de 2-cétobutyrate, avec les conséquences remarquables indiquées plus haut. On a là sans doute la raison de l'effet observé lors du passage d'une croissance anaérobie à une croissance en présence d'oxygène. En effet la respiration se trouvant brusquement augmentée, l'ensemble du flux de la glycolyse se trouve détourné vers le cycle de Krebs, ce qui implique une mobilisation massive de la pyruvate déshydrogénase et par conséquent, une carence en substrat des acétolactate-synthases...
Ces résultats nous ont amenés à chercher si le 2-cétobutyrate ne serait pas une "alarmone" destinée à réorganiser le métabolisme et l'expression génétique lors du passage de l'anaérobiose à l'aérobiose. Et nous avons montré à la fois biochimiquement et génétiquement que tel est effectivement le cas. On observe au cours du passage de l'anaérobiose à l'aérobiose une synthèse massive du cétoacide. Ce cétoacide serait ainsi l'un des éléments de l'effet Pasteur. On remarquera par ailleurs que comme l'aspartate est le précurseur ultime du cétobutyrate la régulation que nous avons découverte fournit la rétroaction qui manquait et qui évite qu'en effet on n'arrive à une concentration explosive du cétoacide.
Nous avons choisi dans le contexte des programmes de séquençage de génomes entiers de créer un système d'expertise informatique permettant la consultation automatique d'une base de connaissance sur le métabolisme, liée à la base de données des gènes, que nous avons constituée par ailleurs (thèse de Thierry Rouxel). La base de données correspondante, Metalgen, a été présentée dès le 2ème Congrès International sur le Génome de E. coli qui s'est tenu à Madison en septembre 1993 (et dont j'étais l'un des organisateurs). Elle a été poursuivie par le développement d'une nouvelle base de données, fondée sur l'exploration du concept de "voisinage", Indigo, en collaboration avec P. Nitschké et A. Hénaut de l'Université de Versailles Saint-Quentin. Cette approche est à l'origine du concept de "voisinages" essentiel pour le développement de la génomique fonctionnelle, et développé ensuite un peu partout dans le monde. T. Rouxel a développé par ailleurs de nouvelles méthodes d'évaluation des bilans de matière et des bilans énergétiques en fonction des chemins métaboliques employés. A la suite de l'intérêt manifesté par la société ORSAN ces travaux se se sont développés sous la forme d'une collaboration nationale avec des laboratoires de Clermont-Ferrand, Nancy, Bordeaux et Versailles. Ils ont été financés par le programme "usine cellulaire" du 4e programme cadre de l'Union Européenne. Plus récemment ils ont permis la création d'une compagnie de service pour l'analyse du métabolisme au sein de la technopole de Clermont-Ferrand (Metabolic Explorer) après que la compagnie ORSAN a été rachetée par un industriel japonais.
Les travaux décrits jusqu'ici indiquent que le métabolisme intermédiaire — sujet d'intérêt majeur de la biologie, aujourd'hui mal considéré mais peu à peu réhabilité — joue un rôle de premier plan dans la régulation de l'expression génétique. La synthèse des petites molécules est d'un ordre conceptuel différent mais aussi importante que celle des macromolécules. Et il est nécessaire, si l'on cherche à retourner aux questions d'origine (à celle des origines de la vie, en particulier), de considérer que l'évolution de ce métabolisme particulier est inséparable de l'évolution du métabolisme des macromolécules. Cela m'a amené à prendre en compte les problèmes de l'origine du métabolisme intermédiaire. Dit autrement, il est impossible de se pencher sur les problèmes des origines de la vie à partir de considérations sur les seules macromolécules, que leur naissance et leur évolution a dû se faire de façon concomitante avec la naissance et l'évolution de la biosynthèse des petites molécules.
Plusieurs résultats du laboratoire ont conforté cette idée, et m'ont conduit à une réflexion sur l'intérêt de connaître la séquence de génomes entiers. Par exemple nous avons découvert (fortuitement) au cours de l'analyse de la région amont de l'opéron ptsHptsIcrr du colibacille, un gène de synthèse de la cystéine (cysK) dont le produit est très voisin d'une sous unité de la tryptophane synthase. Cela relie la synthèse de la cystéine et du tryptophane, acides aminés dont les codons sont dans la même case du tableau du code génétique. Ainsi peut-on s'interroger sur l'origine, et l'ancienneté des métabolismes correspondants. Plusieurs travaux sur la structure des nucléotide kinases, nous ont montré que l'idée de Granick, que les activités enzymatiques les plus anciennes se sont spécifiées de plus en plus au cours du temps, représentait très probablement ce qui s'est produit au cours de l'évolution prébiotique, et de l'évolution des premières cellules. Cela donnait tout son sens à un programme de recherche qui considérerait en priorité l'exploration et la comparaison des génomes bactériens en entier. Et cela nous donnait un premier élément d'intérêt pour le métabolisme du soufre.
Avant d'impliquer mon laboratoire dans un projet aussi considérable que celui du séquençage d'un génome entier j'ai sollicité l'avis d'un certain nombre d'experts. D'abord, au CNRS fin 1986, j'ai consulté les représentants des différentes sections du Comité National impliqués dans la rédaction du Rapport de Prospective. A mon grand plaisir, j'ai reçu un encouragement tout à fait significatif de la part de certains de mes confrères. Cela m'a incité à aller plus loin, et à tester les réactions de la communauté des microbiologistes, au cours de la conférence inaugurale de la réunion organisée par la Société Française de Microbiologie en mars 1987. Là encore, malgré les très grandes réticences de beaucoup, l'intérêt pour ce projet s'est manifesté et j'ai pu commencer à discuter des contraintes pratiques avec Simon Wain-Hobson, expert reconnu des techniques (et des difficultés) du séquençage à grande échelle. A la fin du printemps 1987 Jim Hoch, aux états Unis, proposait au cours d'une réunion internationale, à ses partenaires européens, et en particulier à Raymond Dedonder, d'entreprendre en commun, la détermination de la séquence complète du génome de Bacillus subtilis. Il devenait donc naturel que les efforts soient associés dans un projet de séquençage de grande ampleur, le cas de B. subtilis paraissant particulièrement bien adapté. Nous avons donc convenu d'une association entre les unités de R. Dedonder et la mienne pour cette entreprise.
Ce choix et la stratégie définie sont détaillés dans mon livre La Barque de Delphes, aux Éditions Odile Jacob en mai 1998, et réédité, sous une forme nouvelle à Harvard University Press en 2003, sous le titre The Delphic Boat. La littérature a exposé les possibilités d'atteindre la connaissance complète du génome humain au travers de la détermination de la séquence des nucléotides qui constituent les chromosomes. Mais peu a été dit sur les raisons fondamentales qui concouraient à donner le plus grand intérêt aux projets de ce type.
L 'autonomie des êtres vivants suppose l'existence d'une cohérence interne de leur génome. L'ensemble des règles fixées par la séquence de l'ADN —qui prend son sens non seulement localement mais encore sur de grandes distances— suffit à décider de la survie et de la reproduction, et le programme correspondant est de taille finie. Les règles de réécriture du patrimoine génétique (transcription et traduction), qui imposent, à partir de la mémoire que constitue l'ADN, la structure des effecteurs du métabolisme, sont totalement incluses dans la suite des nucléotides et des acides aminés qui leur correspondent. Notons cependant que cela ne signifie pas qu'il n'existe pas des contraintes essentielles en dehors de l'enchaînement des nucléotides et des acides aminés. De même, il est essentiel de remarquer que, loin de se comporter comme une simple suite de nucléotides enchaînés que l'on pourrait comparer à une suite au hasard, tout ADN reflète le développement d'une histoire qui a pris en compte toutes sortes de contraintes et d'explorations de l'environnement. Jusqu'à cette époque, l'aspect cohérent de l'information correspondante, fruit de l'histoire évolutive, était restée inaccessible, et l'on n'était pas vraiment en mesure d'identifier la nature des signaux qui dictent la mise en place diachronique ou architecturale des macromolécules conservant et exprimant le programme génétique. Grâce aux techniques de séquençage de l'ADN, il devenait de plus en plus facile d'avoir accès à la structure exacte totale d'un organisme, et donc d'avoir accès à cette cohérence.
Dans la mesure où les projets de séquençage supposent beaucoup de travail, le choix des organismes a eu un rôle crucial. Des considérations politiques et économiques, plus que scientifiques, ont guidé ces choix. Il aurait été naturel d'avoir une idée de la façon dont serait exploitée l'information que véhiculent ces génomes, et, par conséquent, d'avoir une idée de la nature de cette information dans un génome donné et de privilégier la recherche correspondante. C'est pourquoi j'ai exploré, en collaboration avec un informaticien, Olivier Gascuel, le développement de méthodes nouvelles d'analyse des séquences (nucléotidiques ou polypeptidiques) pour en extraire des descripteurs. Cela nous a conduits à proposer un descripteur spécifique des peptides signaux chez les entérobactéries. Devant les succès correspondants, j'ai entrepris de fédérer, avec Alain Hénaut et Alain Viari (alors à l'Institut Curie et qui a longtemps animé l'Atelier de Bioinformatique, structure informelle créée par Henri Soldano en 1985), un groupe de travail en intelligence artificielle qui se consacrait au traitement des séquences. Parmi les résultats obtenus, une collaboration avec François Rechenmann de l'IMAG à Grenoble, a permis à Alain Viari et Claudine Médigue de créer un système d'analyse automatique des séquences, ImaGeneTM, et de fournir un ensemble de logiciels d'intelligence artificielle pour le traitement des séquences. Nous étions donc prêts à envisager sérieusement de participer à grande échelle à la détermination de la séquence totale du génome d'un organisme. Cette association a été confortée de façon formelle par la création pour 4 ans (1992-1995) d'un GDR du CNRS dont j'ai été le directeur avec François Rechenmann (GDR 1029 : Rapport final 1995). Ce GDR a mené son activité en concertation avec le GIP GREG, dont le comité sectoriel "informatique et génomes" que je présidais, a permis d'organiser de façon harmonieuse les efforts français de recherche en informatique.
Visant la visibilité médiatique, les projets de séquençage génomique ont surtout mis l'accent sur le génome humain. En dehors du problème de la très grande taille de ce génome (3,5 milliards de paires de bases), les génomes eucaryotes supérieurs contiennent une charge d'ambiguïtés considérable dues à la façon dont l'évolution a procédé. Au moins 95% de la séquence de ces génomes correspond des traces du passé qui, parce qu'elles ne représentent pas une "charge" trop lourde, et souvent parce qu'elles ont acquis une fonction au cours de l'évolution (comme "espaceurs" ou "minuteurs"), sont conservées au cours des temps. D'autres raisons, impliquant divers mécanismes assurant la stabilité de ce génome, ont aussi pour conséquence de produire un important polymorphisme dans les copies qui ne sont pas directement responsables de l'expression génétique (c'est-à-dire de la synthèse des molécules qui seront les effecteurs de la vie cellulaire). Ainsi, ce qui n'est pas un encombrement rédhibitoire pour un génome l'est sans aucun doute pour commencer l'exploitation informatique. Il était donc essentiel, pour mettre en place un projet raisonnable, de choisir d'abord des organismes dont le génome est aussi compact que possible ou, dans l'idée d'explorer le génome humain dans son ensemble, dont la cohérence soit biologiquement significative. Ce n'est pourtant pas ce qui a été fait initialement.
Ce sont les bactéries qui se prêtent le mieux à l'étude des génomes. Leur génome est compact, et nous disposons de beaucoup d'information à leur sujet. Elles ont un intérêt évident pour l'environnement, l'industrie ou la médecine. Enfin, elles sont manipulées aisément, ce qui permet d'exploiter rapidement les connaissances obtenues. La génétique moléculaire s'est élaborée à partir de leur étude, et c'est donc à partir des modèles qui ont constitué cette science que se sont organisés les projets de séquençage. Cela explique que nous assistons, depuis 1998, à la publication d'abord d'un nouveau génome entier chaque mois, puis, en 2008 plusieurs dizaines et désormais des milliers. Le choix d'une bactérie particulière était difficile, et mettait en jeu, en dehors des considérations scientifiques, des considérations politiques. L'organisme le plus prometteur était E. coli. Kohara, Isono et leurs collègues avaient en effet publié une carte de restriction totale de l'ADN (4720 kpb, donc sensiblement plus long qu'on ne le pensait initialement). Annoncé par Georges Church comme presque terminé en 1988, le séquençage du génome de E. coli devait en réalité être terminé mi-1997 après de nombreuses péripéties. Nous avons choisi, pour calibrer nos techniques informatiques d'exploitation des séquences, de constituer une base de données formée de la mosaïque des gènes de cet organisme isolés un peu partout dans le monde. Cette base spécialisée, Colibri, construite par Claudine Médigue, et mise à jour par Ivan Moszer, rendait l'information utilisable pour le génome du colibacille.
Si l'on sépare l'ensemble des gènes bactériens en deux catégories, on peut, grossièrement, estimer qu'une fraction correspond à la perennisation et la survie, le reste à l'occupation d'une niche écologique spécifique. Nos travaux (2005-2008) ont donné corps à cette hypothèse en identifiant avec une grande précision les gènes qui forment le cœur du génome (le paléome) et ceux qui contribuent à l'occupation d'une niche (le cénome). Les entérobactéries (dont E. coli fait partie) gèrent bien le passage aérobiose/anaérobiose; les Pseudomonas, le catabolisme; beaucoup de Gram positifs, la sporulation; les algues bleues, la photosynthèse; les actinomycètes, le métabolisme secondaire; les myxobactéries, la différenciation; les archébactéries, toutes sortes de milieux extrêmes... Nous connaissons la séquence totale du génome d'un type de chacune de ces classes, et cela permet de mieux comprendre avec précision ce qui permet l'occupation de la niche correspondante. Techniquement cependant, le premier choix a été orienté d'abord vers les génomes les plus petits, et aussi riches en A et T que possible, afin d'éviter un certain nombre de problèmes d'identification de phases de lecture du texte génénomique, et aussi certaines difficultés de manipulation au cours du séquençage. C'est cette très petite taille, au delà des considérations politiques, qui explique que les deux premiers génomes connus aient été ceux de Hæmophilus influenzæ et Mycoplasma genitalium, déterminés en 1995, par Craig Venter et ses collègues à un moment où l'on connaissait déjà la séquence de fragments continus beaucoup plus longs chez Bacillus subtilis.
Ces organismes ont évolué vers la pathogénicité en perdant bien des fonctions qu'avaient leurs ancêtres et il était donc utile d'entreprendre le séquençage d'autres organismes, beaucoup plus autonomes. Un organisme bien connu, modèle de la sporulation, répondait à la question, c'est Bacillus subtilis. Cette bactérie est aussi d'un intérêt industriel évident (elle est productrice d'enzymes très utilisées). Par ailleurs, comme de nombreuses bactéries de cette famille sont pathogènes, il s'agit d'un modèle utile à explorer à des fins médicales. La génétique de B. subtilis était moins bien connue que celle de E. coli. Mais en 1994 plus de 800 gènes avaient été localisés sur le chromosome, dont la longueur est un peu plus courte que celle du chromosome de E. coli (environ 4200 kpb). Un ensemble concerté de laboratoires aux Etats-Unis, au Japon et en Europe collaborait depuis de nombreuses années à l'étude de certaines fonctions essentielles de cette bactérie : compétence, sécrétion des protéines, réplication, recombinaison, sporulation, germination... Son génome est relativement riche en A+T, ce qui facilite, au cours du séquençage, le repérage des erreurs par insertion ou délétion. Mais la souche de laboratoire de B. subtilis avait surtout l'avantage d'être transformable (c'est d'ailleurs une bactérie voisine qui a permis la découverte du rôle de l'ADN dans l'hérédité). Il était donc possible d'y introduire à volonté de l'ADN modifié in vitro. Comme les mécanismes de recombinaison permettent très efficacement la recombinaison au site homologue, la génétique inverse s'y pratique de façon routinière (elle était alors plus aisée que dans E. coli). Il était alors facile de modifier tout gène dont on soupçonne l'intérêt et de mesurer les effets de cette modification in vivo. La découverte de gènes inconnus pouvait donc précéder l'exploration de leur fonction dans la physiologie bactérienne, à la différence de ce qui se produit habituellement.
Toutes ces raisons expliquent que nous ayons entrepris, en collaboration avec l'Unité de Biochimie Microbienne qui a assuré la coordination administrative du projet (R. Dedonder, puis Frank Kunst), et quatre laboratoires européens initialement, de séquencer une partie de ce génome. La collaboration s'est poursuivie par l'entrée en scène du Japon (qui a séquencé un tiers du génome) et d'un grand nombre d'autres laboratoires européens. Outre notre participation directe à l'effort de séquençage proprement dit nous assurons la coordination de l'analyse et de la gestion des données : le génome a été entièrement reséquencé en 2007 et je l'ai entièrement réannoté, au cours de plusieurs cycles, le dernier en 2018. L'effort de réannotation se poursuit. Dans un premier temps nos travaux ont été soutenus par le programme SCIENCE de la CEE. Cependant, de 1991 à 1993, nous n'avons pas obtenu de soutien CEE, mais nous avons été aidés par la direction de l'Institut Pasteur, et par un crédit du Ministère de la Recherche. A partir de 1994, nous avons obtenu un soutien du programme BIOTECH de l'Union Européenne, renouvelé jusqu'à fin 1998. La séquence totale du génome à été publiée en novembre 1997. Elle était alors la dixième, mais est restée très longtemps la seule d'un Firmicute de cette taille (le génome de Bacillus anthracis n'a été connu qu'en 2003 ! alors que plus de 300 génomes avaient alors été séquencés).
Pour cette entreprise nous avons créé un laboratoire de séquençage et organisé sous forme semi-automatisée (les réactions de séquence étaient robotisées) la collaboration d'un ensemble de deux à cinq étudiants, chercheurs ou techniciens (suivant les moments), venant d'horizons variés pour déterminer le plus efficacement possible la séquence de grands fragments d'ADN, et pour faire ensuite la génétique inverse des gènes intéressants. En dehors des problèmes classiques posés par le séquençage à grande échelle, B. subtilis a créé un problème particulier car son ADN est très toxique dans E. coli (cela est dû, nous le savons aujourd'hui, à ce que les signaux de traduction et de transcription chez B. subtilis sont extrêmement forts chez E. coli). Nous avons entrepris l'étude de certains par génétique inverse et caractérisé les conditions de leur expression. Nous avons ensuite concentré nos efforts sur l'élucidation des chemins métaboliques de molécules ubiquistes, mais à la fonction qui reste énigmatique, comme les polyamines. Cela nous a conduits à nous intéresser à un métabolisme relié, le métabolisme du soufre, dont l'importance paraît si grande qu'on comprend mal pourquoi il a été si peu l'objet d'études détaillées. Et cela a été pour nous une surprise que de découvrir que des pans entiers de notre connaissance du métabolisme intermédiaire restaient complètement inconnus, et valaient donc la peine d'une exploration approfondie.
La séquence complète du génome était connue en avril et rendue publique en novembre 1997, après que les contrôles de qualité appropriés eurent été faits. Comme dans le cas de la levure, le séquençage "au hasard" de fragments du génome de B. subtilis nous avait révélé dès 1991 l'existence d'un très grand nombre de gènes (la moitié de ceux qui ont été identifiés) dont le produit ne s'apparente à aucun gène connu. Il s'agit de la première découverte de la génomique, qui démontre que les approches génétiques classiques ont laissé de côté un pan immense de notre connaissance des organismes, dont nous n'aurions même pas soupçonné l'existence si nous n'avions commencé à mettre en place des projets de séquençage de grands génomes. Cela seul suffisait donc à justifier de tels projets.
À la même date une autre grande première fut la découverte de l'importance du transfert génétique horizontal dans la genèse et le maintien des génomes. Jusqu'à cette date on savait bien que les génomes contenaient souvent des provirus intégrés (prophages dans le cas des bactéries) en leur sein, mais cela paraissait anecdotique. Nous avons montré que cela n'était que la partie émergée d'un immense iceberg : les génomes contiennent une fraction très importante de gènes étrangers, d'abord accommodés ou tolérés, puis peu à peu assimilés sous la forme de gènes peu distincts des gènes plus anciens. Cette découverte est restée ignorée jusque vers les années 2000 où une publication dans un journal à la mode a répandu l'idée et s'est peu à peu substituée à la découverte elle-même utilisant en particulier une nouvelle expression (transfert génétique latéral) pour l'occulter ! Ce processus de piratage des découvertes a été bien décrit par Jacques Ninio.
Dans la mesure où la séquence publiée rassemblait les travaux de plus de 30 laboratoires, il nous a paru essentiel, en 2007, de séquencer à nouveau le génome d'un clone de la bactérie, en collaboration avec le Genoscope. Ces travaux nous ont montré l'existence de plus de 2000 variations, essentiellement dues à des erreurs de séquence.
En dehors de l'expérimentation in vivo, l'analyse des séquences au moyen de techniques informatiques permet d'explorer bien des questions par une expérimentation "in silico" : description des signaux collectifs, traits essentiels d'un gène ou d'une protéine, parenté phylogénétique.
L'accent sur cet aspect a été mis en avant en 1988-1989 au cours des réunions de coordination qui ont conduit à la création du projet de séquençage du génome de B. subtilis, pour lesquelles l'expression "in silico" a été créée. Plusieurs niveaux bien identifiables sont à distinguer. D'une part, il faut considérer l'apport informatique à l'acquisition des données, d'autre part la nécessité d'en comprendre au mieux le sens biologique, enfin il est nécessaire de gérer les données et les connaissances associées. Ces aspects sont reliés par le fait que la vérification de la validité des séquences acquises peut se faire grâce à la mesure de leur cohérence biologique. Par exemple, si l'on crée des descripteurs efficaces de ce qu'est une phase codante (CDS), toute erreur par insertion ou délétion d'une base (ce qui se rencontre assez fréquemment en cours d'acquisition) sera immédiatement repérée et soumise à un processus de correction qu'il conviendra de mettre en place. L'approche informatique doit être à la fois intégrée et dynamique. C'est inhabituel et peu compatible avec l'utilisation classique des modèles algorithmiques. Il est essentiel d'autre part d'impliquer la communauté des expérimentateurs afin d'éviter l'exploration stérile d'hypothèses qui n'ont rien à voir avec la biologie, et, bien sûr d'orienter l'exploration vers des questions d'intérêt biologique. Les bases de connaissance utilisées sont évolutives et fonctionnelles, ce qui est du domaine de la recherche en Intelligence Artificielle en particulier. Cela m'a conduit à proposer au CNRS la constitution d'une structure de recherche, permettant la collaboration entre informaticiens et généticiens dans le domaine du traitement des séquences. Cette proposition a été acceptée au sein du programme IMABIO, et a conduit à la constitution au début de l'année 1992 d'un GDR (groupement de recherche) associant une cinquantaine de chercheurs, presque tous informaticiens, rassemblés dans un programme Génomes et Informatique, et dont j'ai été le directeur. Une idée-force de ce GDR était de faire traiter les mêmes données (en particulier celles qui sont regroupées dans les bases de données spécialisées que nous créons) par les différents groupes, de façon à les mettre en perspective sous des éclairages différents. Le bilan de quatre années de recherches de ce GDR, au cours d'une réunion organisée mi-octobre 1995 a bien mis en évidence l'effet bénéfique de ces collaborations (GDR 1029). Plusieurs associations stables entre chercheurs des deux disciplines en sont issues.
Les fragments obtenus au cours du séquençage sont explorés pour y reconnaître des signaux pertinents. C'est à la genèse de descripteurs de ces signaux que s'emploie une part importante de la recherche en bioinformatique. Pour cela, on utilise des techniques d'apprentissage par assimilation ou par discrimination, mais aussi des techniques plus classiques du traitement du signal ou de l'analyse des données. Par ailleurs, les logiciels doivent être évolutifs de façon à s'affiner au fur et à mesure qu'augmente le nombre des séquences obtenues. Nous avons constitué deux banques de données de référence (contenant les données brutes) traitée sous la forme de bases de données (évolutives et contenant les commentaires et le traitement partiel) pour E. coli (Colibri) et B. subtilis (SubtiList). Ces bases de données sont disponibles pour l'ensemble de la communauté internationale sur le réseau Internet. Un nouvel ensemble de bases de données construites sur le même principe (mais gérées par des Logiciels Libres) a été mis en place au HKU-Pasteur Research Centre que j'ai créé à Hong Kong.
Un aspect important de l'annotation est la comparaison des séquences obtenues avec ce qui est connu dans les banques internationales (processus dont les premiers éléments devraient être quasi automatiques) puis la constitution d'arbres de relations, permettant, entre autres choses, une évaluation des parentés phylogénétiques. C'est d'ailleurs la découverte d'une parenté inattendue (entre la synthèse de la cystéine et celle du tryptophane) qui nous a renforcé dans notre conviction de l'intérêt des projets de séquençage des génomes. A ce stade, il se constitue des familles de produits de gènes apparentés et il convient d'apprécier de façon fine la nature de ces parentés par la construction de logiciels de comparaison floue. Il faut noter ici, qu'en l'absence de données expérimentales in vivo ou in vitro l'annotation ne saurait être considérée comme permettant l'identification fonctionnelle définitive d'un gène. Il ne s'agit là que d'un pas préliminaire dans la direction de cette identification.
Les projets de séquençage supposent l'existence d'une cohérence interne : il doit donc exister des signaux collectifs ou des "absences" collectives. Une analyse statistique fine (analyses multivariées) des gènes et de leurs produits permet de repérer ces signaux. Par apprentissage, on peut ensuite en créer les descripteurs (qui seront réinjectés dans les programmes d'acquisition, au moins pour certains d'entre eux). Ces descripteurs seront alors confrontés aux données biochimiques, physico-chimiques ou physiologiques que l'on pourra posséder sur l'organisme étudié. Par ailleurs, on recherche une corrélation entre classes statistiques et classes d'activités. Un apport intéressant est d'associer les gènes aux réactions connues du métabolisme intermédiaire (un à-côté du séquençage du génome de B. subtilis est donc la constitution de la base de données "métabolisme", liée à la base de données "gènes et produits de gènes"). Ce travail, commencé en collaboration avec Alain Hénaut (et, initialement, la Société ORSAN) par la création de la base Metalgen, puis développée au travers du serveur Indigo jusqu’à la disparition du Centre National de Ressources Infobiogen, s'est poursuivi par une réflexion menée dans un programme de recherche coordonnée financé par l'European Science Foundation qui a donné lieu au Réseau d’Excellence Européen BioSapiens. Il a par ailleurs permis la naissance de la société Metabolic Explorer.
Les programmes de séquençage ont montré que beaucoup de gènes sont inconnus et ne conduisent pas à des phénotypes aisément repérables, et l'un des apports majeurs de cette exploitation informatique est de proposer des hypothèses sur leurs fonctions physiologiques. L'analyse factorielle des correspondances de l'ensemble des gènes les classe sans ambiguïté en trois classes et non deux, comme on le pensait jusqu'alors. Les deux premières correspondent, d'une part, aux gènes centraux du métabolisme cellulaire : traduction et transcription, cœur du métabolisme intermédiaire, contrôle du repliement des protéines, et, d'autre part, aux gènes responsables de la plupart des biosynthèses des petites molécules, et d'une façon générale exprimés peu et souvent ou rarement et parfois fortement. La troisième classe rassemble des gènes qui ont en commun le fait qu'ils peuvent participer aux échanges génétiques horizontaux (HGT; récepteurs de phages, réplication de virus ou de plasmides, transposition). Ce qui est remarquable, c'est que l'usage du code génétique est chez ces gènes très particulier. Ce qui l'est encore plus est que les gènes cdoant les protéines permettant la fidélité de la réplication en font partie : cela suggère que les transferts horizontaux jouent un rôle spécifique dans la spéciation, au moins des entérobactéries (thèse de Claudine Médigue). Ce travail était la première démonstration de l’importance du transfert génétique horizontal chez les bactéries. Il a été poursuivi par une étude comparable chez B. subtilis avec des résultats semblables. Elle correspond à une anomalie de l'usage du code qui est fortement enrichi en A + T. Une étude fine du biais d'usage des codons a montré plus récemment (2006) qu’il se forme des îlots d’usage semblable dans le chromosome, et que la traduction est donc au cœur de son organisation architecturale.
Ces résultats confirment l'intérêt —la nécessité scientifique— des programmes "génomes". La génomique a créé une génétique nouvelle, par exemple liée aux phénomènes transitoires nécessitant des horloges. Elle a mis en évidence l'existence de gènes totalement inconnus et imprévus. Elle a conduit à s'interroger sur la nature des fonctions biologiques, et en particulier sur les liens qui les relient à des structures : le plus souvent la structure ne dit pas la fonction, mais c'est l'inverse, ce sont les fonctions qui capturent des structures. Des relations phylogénétiques intéressantes ouvrent de nouvelles perspectives pour comprendre l'évolution. Il est possible enfin d'espérer comprendre ce qu'est le cœur d'un être vivant, et de porter de nouvelles questions et de nouvelles réponses sur les origines de la vie.
Nous avons développé plusieurs bases de données pour gérer la connaissance biologique sur les génomes de E. coli et B. subtilis. Il fallait aller beaucoup plus loin. Nous avons donc, en partant de l'infrastructure du GDR, organisé la construction d'un environnement capable de gérer non seulement la connaissance factuelle sur les séquences, mais la connaissance méthodologique associée. Plusieurs membres du groupe de travail "Modélisation des connaissances" au sein du GDR sont à l'origine d'un projet soutenu par le GIP GREG (Groupement de Recherches et d'études sur les Génomes) qui a donné lieu à la plateforme ImaGeneTM, en beta-test en juin 1998 et aujourd'hui commercialisée sous le nom de Genostar par la société IOGMA.
Ce projet a réalisé un système interactif d'aide à l'analyse de séquences qui permet :
. de mettre en oeuvre aisément des méthodes d'analyse ;
. d'aider un utilisateur à choisir la ou les méthodes adéquates pour une tâche donnée et à enchaîner des méthodes entre elles dans le cas de tâches plus complexes ;
. de mémoriser et de gérer à la fois les données de l'analyse et les résultats produits par l'application de méthodes ;
. d'étendre le système en intégrant de nouvelles méthodes et leurs modes d'emploi.
Les méthodes d'analyse de séquences sont décrites en termes des objets qu'elles admettent en entrée et qu'elles produisent en sortie. La description de ces classes d'objets fait partie intégrante de la base de connaissances. Les tâches permettent de décrire des enchaînements adaptatifs de méthodes : la décomposition d'une tâche en sous-tâches plus simples dépend des entrées de la tâche. Les connaissances apportées par les tâches sont ainsi d'ordre méthodologique : elles permettent au système d'aider l'utilisateur dans le choix et l'enchaînement de méthodes pour la résolution d'un problème donné, comme la recherche de zones codantes.
![]()
Figure 1 - Pour résoudre un problème d'analyse, ce dernier est décomposé de façon récursive en sous-problèmes jusqu'à l'obtention de problèmes suffisamment élémentaires pour être résolus directement par l'exécution d'une méthode. à tout niveau, la décomposition d'un problème en sous-problèmes est opportuniste, en ce sens qu'elle dépend des entités sur lesquelles portent ce problème, entités qui résultent elles-mêmes de la résolution antérieure d'autres problèmes. Le choix de la décomposition adaptée résulte d'une phase de caractérisation, par classification hiérarchique, du problème courant. Dans le modèle de connaissances développé, les problèmes et sous-problèmes sont représentés par des tâches et des sous-tâches reliées par des opérateurs de classification et de décomposition.
Une interface cartographique a été aussi développée, destinée à représenter et à interagir avec les données de séquences et les résultats des analyses. Compte tenu de la diversité des objets impliqués et de leur représentations possibles, les spécifications de cette interface ont mis l'accent sur sa généricité. Les utilisateurs ont ainsi la possibilité de composer leur écran en affichant plusieurs cartes différentes (fig. 2), en ajustant leurs tailles, en sélectionnant les classes d'objets à y faire apparaître, en spécifiant les échelles et leur unité, en choisissant, voire en définissant, les icônes associées aux différentes catégories d'objets, etc.
![]()
Figure 2 - L'interface permet de visualiser simultanément plusieurs cartes de natures (types des objets) et d'échelles différentes. L'utilisateur a la possibilité de modifier ces caractéristiques et de se constituer ainsi une interface bien adaptée à la classe de problèmes qui l'intéresse.
En plus de fonctionnalités classiques, telles que le "zoom", l'interface offre plusieurs fonctionnalités avancées. Il est ainsi possible d'établir des liens entre plusieurs cartes et de synchroniser le défilement des objets dans la fenêtre de visualisation en tenant compte de l'existence de ces liens (fig. 3). Il est également possible d'introduire, pour une même carte, plusieurs zones d'affichage. Typiquement, des informations associées aux 3 phases de lecture sur les deux brins d'ADN peuvent ainsi être très clairement représentées. Enfin, l'utilisateur peut rapidement faire disparaître et réapparaître sur une carte des classes d'objets particulières.
![]()
Figure 3 - Exemple de liens entre cartes. Les objets de type gène de la carte génétique du haut de la fenêtre sont liés aux objets dénotant les mêmes gènes sur la carte physique du bas. L'interface offre plusieurs modes de défilement synchronisé des deux cartes en tenant compte de ces liens.
L'ensemble de ces travaux a donné lieu en 2005 à la création de la plateforme d'annotation MaGe par Claudine Médigue et ses collègues, au Génoscope, Centre National de Séquençage.
Outre les bases de données spécialisées mentionnées plus haut, nous avons obtenu d'intéressants résultats sur l'organisation des chromosomes. Notre découverte que le chromosome de E. coli est une mosaïque de quatre classes de gènes, une classe étant particulièrement impliquée dans les échanges génétiques horizontaux (entre espèces) nous a permis, en collaboration avec M. Borodovsky (Georgia Tech) d'affiner la prédiction des zones codantes dans cet organisme, de l'étendre à B. subtilis, puis de montrer que cette répartition des biais d'usage des codons correspondait à une organisation fine du chromosome par la traduction. L'analyse des classes de gènes et l'étude des mots dans les séquences nous ont conduit à faire de nombreuses observations inattendues. L'analyse des propriétés du tétranucléotide GATC, chez E. coli, met en évidence une répartition à longue distance (1100 bp) de ces sites, ce qui correspond au système de réparation des mésappariements. Elle indique une forte contre sélection des sites de liaison de la protéine CAP et elle prédit enfin l'existence d'un contrôle spécifique du passage de l'anaérobiose à l'aérobiose, par un facteur reconnaissant ce tétranucléotide. De même nous avons prédit l'existence d'un mécanisme de "glissement" de la lecture de certains ARN messagers chez E. coli et B. subtilis, au voisinage de tétranucléotides AGCT, permettant une expression épigénétique de plusieurs produits à partir d'un même gène.
Plus généralement, l'analyse de l'organisation des gènes le long du chromosome indique qu'elle n'est pas au hasard, comme on pourrait le penser superficiellement, mais qu'elle est corrélée à la position de leur produit dans la cellule. En quelque sorte le plan de la cellule est dans le chromosome.
Enfin, un résultat particulièrement intéressant est sorti de l'analyse comparative des génomes de E. coli et B. subtilis. En bref, très peu de régions sont conservées entre ces deux organismes, et une région particulière, formant chez E. coli un opéron entre le gène cmk (cytidylate kinase) et rpsA (protéine ribosomique S1) est conservé chez B. subtilis. L'analyse de cette structure, et de la structure du métabolisme intermédiaire chez les bactéries, montre alors que, si l'on s'en tient à ce qui est connu, la synthèse de novo d'ADN est ... impossible ! Il manque en effet un précurseur essentiel, le CDP. En résumé, l'analyse démontre que c'est par le biais de la dégradation des ARN messagers, et la rotation des phospholipides que cette molécule indispensable est synthétisée. Chez les eubactéries la polynucléotide phosphorylase produit directement le CDP, et tout conduit à penser que la protéine S1 (ou son analogue chez B. subtilis) joue un rôle de premier plan dans ce mécanisme dégradatif. Ainsi, la véritable fonction de la polynucléotide phosphorylase serait de produire assez de CDP pour permettre une synthèse adaptée d'ADN.
D'une façon générale on en trouvera un résumé dans le chapitre de la "bible" des spécialistes de E. coli Escherichia coli and Salmonella typhimurium, Cellular and Molecular Biology (ASM Press), paru en 1996 où nous exposons les apports principaux de l'analyse in silico chez cet organisme. Il n'est pas possible ici de faire part de l'étendue inespérée des résultats glanés par l'analyse des génomes complets. On pourra en trouver un résumé dans mon livre La Barque de Delphes, ou ce que révèle le texte des génomes (Odile Jacob, 1998) adapté et mis à jour pour un public anglo-américain, The Delphic Boat.
Les analyses génomiques in silico sont désormais complémentaires des analyses in vivo et in vitro, et préfigurent une génétique entièrement nouvelle, fascinante, à l'aube du troisième millénaire.
![]()