De la conoissance et merveilles du monde et de
l'homme
Pierre de DAMPMARTIN
Génomique
Genèse de la
génomique 1988 (en anglais)
In silico 1989
Le GDR 1029
Plaidoyer 1996
Bases de données
Notre participation
Le génome humain
Le vaisseau de Thésée
La cascade Toll
Glossaire
Monographies ![]()
The
methionine salvage pathway
Sulfur metabolism
The H-NS regulator
Le grand séquençage des « petits » génomes
Comment définir complètement la composition d'un être vivant en termes chimiques ?
Depuis plusieurs années on entend parler, sous la forme d'informations généralement plus médiatiques que scientifiques, d'un grand et bien énigmatique projet proposé par les biologistes - et l'on mentionne surtout les chercheurs américains - qui consisterait à "déterminer la séquence complète du génome humain". Et d'aucuns d'invoquer les projets collectifs des grands appareils de physique, ou encore les pas de l'Homme sur la Lune… A travers ce discours qui tendrait à réduire l'être humain à une mécanique et à valoriser un exploit technique est-il possible d'entrevoir un réel projet scientifique ? Je vais tenter ici de répondre par l'affirmative, en posant la question autrement qu'en ces adjectifs emphatiques qui font bien souvent la une des journaux, et en montrant tout ce que peut apporter déjà le séquençage de génomes de différents organismes. Mais avant d'entrer dans le vif du sujet, et afin de placer les projets de séquençage de génomes entiers dans leur contexte biologique je vais d'abord revenir brièvement sur une question difficile, mais essentielle : qu'est-ce que la vie ?
Quatre processus et une loi centrale sont nécessaires pour définir un être vivant. Les deux premiers concernent principalement les relations entre des petites molécules (formées de moins de cinquante atomes). Le premier (compartimentation) correspond à la formation de cellules ayant un intérieur et un extérieur ; et le second (métabolisme) aux transformations chimiques qui permettent l'édification des cellules. Ces deux processus, qui ont trait à l'organisation et à la nature des flux de matière chez les êtres vivants, sont généralement oubliés de ceux qui décrivent les propriétés de la vie, et, malgré leur importance (ils permettent de comprendre beaucoup de l'origine de la vie) je n'en parlerai ici que très peu. Les deux processus qui sont habituellement retenus, mettent en jeu des molécules géantes, formées de milliers, de millions, voire de milliards d'atomes, et réglent les flux de l'information (mot que je ne puis définir ici, mais dont le flou du sens commun donne une idée suffisante) nécessaires à la vie. L'autonomie d'un être vivant, sa reproduction, son développement et sa survie supposent l'existence d'une mémoire, souvent nommée le "patrimoine génétique", qui se transmet de génération en génération. Ce patrimoine est concrètement représenté par des Acide DésoxyriboNucléiques (ADN), faits de la répétition de quatre motifs chimiquement apparentés, les nucléotides (encore appelés de façon raccourcie, "bases"), selon un enchaînement (la "séquence") semblable à celui d'un texte alphabétique. L'information portée par l'ADN dirige la synthèse des protéines, molécules chimiquement différentes. Celles-ci sont douées de propriétés qu'on peut dire manipulatrices, parce qu'elles permettent les interconversions du métabolisme et les constructions nécessaires à la compartimentation. Ainsi, les réactions du métabolisme sont accélérées et rendues spécifiques (catalysées) par les protéines. Ces dernières forment un enchaînement linéaire, mais de vingt motifs apparentés, les acides aminés, et non plus de quatre. Le fil correspondant se replie dans l'espace de façon à former une architecture spécifique de chaque séquence d'acides aminés, et responsable de la fonction, mais aussi de la localisation des protéines dans la cellule.
La loi biologique centrale consiste en une règle de réécriture qui
permet de passer du texte à quatre lettres de l'ADN au texte à vingt
lettres de la protéine. Cette règle, complexe, se décompose en
plusieurs étapes. La première est une transcription, qui
maintient un alphabet à quatre lettre et consiste à recopier des
fragments de l'ADN, en macromolécules chimiquement voisines, des ARN
(Acide RiboNucléique). Ceux-là, après des modifications appropriées
sont principalement traduits en protéines, en utilisant un
codage qui a chaque suite de trois nucléotides, ou codon, fait
correspondre un acide aminé. Ce code génétique permet,
lorsqu'on possède le texte d'un ADN (sa séquence), de connaître la
séquence de la protéine qui correspond à tel ou tel de ses fragments.
Cela conduit souvent alors prédire la nature, la fonction, la
régulation et la localisation cellulaire de la protéine
correspondante. ![]() C'est là le premier enjeu central des programmes
de séquençage de génomes entiers : il s'agit, en décryptant
le texte de l'ADN, de faire le catalogue de toutes les protéines
codées par le génome considéré, afin de faire l'inventaire de
l'ensemble des fonctions nécessaires à la vie. Le lien avec la
génétique est alors simple, en première approximation, puisqu'à la
suite des travaux d'Ephrussi, Beadle et Tatum, on a établi dans un
grand nombre de cas la correspondance entre gènes — unités formelles
de l'hérédité, conduisant aux caractères observables des individus —
et protéines.
C'est là le premier enjeu central des programmes
de séquençage de génomes entiers : il s'agit, en décryptant
le texte de l'ADN, de faire le catalogue de toutes les protéines
codées par le génome considéré, afin de faire l'inventaire de
l'ensemble des fonctions nécessaires à la vie. Le lien avec la
génétique est alors simple, en première approximation, puisqu'à la
suite des travaux d'Ephrussi, Beadle et Tatum, on a établi dans un
grand nombre de cas la correspondance entre gènes — unités formelles
de l'hérédité, conduisant aux caractères observables des individus —
et protéines.
Le patrimoine génétique spécifie totalement dans la suite des nucléotides, et des acides aminés qui leur correspondent, ce qui est nécessaire à l'édification, au fonctionnement et à la reproduction de chaque organisme. Mais la cohérence de cette information (ce qui fait qu'elle permet le fonctionnement harmonieux de l'organisme), dont une large part résulte de l'histoire évolutive, est encore inaccessible. On n'est pas vraiment en mesure d'identifier la nature des signaux qui dictent la mise en place dans le temps et dans l'espace des macromolécules conservant et exprimant le programme génétique. Et, on le verra, on ne connaît pas - même si on l'a longtemps cru - toutes les grandes lois de la génétique.
Mais revenons aux projets de séquençage. Quelle est l'ampleur du programme de séquençage d'un génome entier ? Que connaît-on déjà ? Les banques de données contiennent plus de 50 millions de bases, provenant des séquences d'ADN d'organismes variés. Mais il s'agit de données presque toujours fragmentaires et hétéroclites. En particulier, en dehors de quelques génomes viraux il est très rare de disposer de séquences correspondant à une zone continue de plus de 50 000 bases (c'est à dire 1% de la longueur d'un génome bactérien). La séquence d'un génome entier serait donc beaucoup plus riche d'enseignements, surtout s'il existe, comme on le pense, des régularités à longue distance dans l'ADN. Cependant, malgré l'amélioration des techniques de séquençage, la lourdeur d'un tel projet impose une grande circonspection dans le choix des organismes. Pour des raisons "spectaculaires" évidentes, et sous l'impulsion de Jim Watson (l'un des inventeurs de la structure de l'ADN, directeur du laboratoire de Cold Spring Harbour, près de New York - fondé en 1927, c'est le plus ancien concerné par la biologie moléculaire), on a surtout fait mention de projets concernant le génome humain : plus de trois milliards de paires de bases réparties en 46 chromosomes, parmi lesquelles 3% environ constituent les 100.000 à 200.000 gènes de l'Homme (*). Cette proportion faible exprime le fait qu'en dehors des gènes, il existe encore de nombreux segments d'ADN qui ne sont que les traces de l'évolution, fixées dans le génome. Ce reste (au moins 95 % chez les mammifères) constitue probablement des "archives", inutilisées actuellement, mais qui, parce qu'elles ne sont pas un trop lourd fardeau, sont transmises héréditairement, car elles fourniront le matériau de base pour la construction de nouveaux gènes, s'il en est besoin.
D'autre part l'ADN est construit de façon modulaire : pour les gènes des protéines, des zones codantes (exons) alternent avec des zones non codantes (introns). Cela se traduit par de nombreuses difficultés. Un ARN messager (intermédiaire transcrit à partir de l'ADN, et spécifiant la synthèse d'une protéine), long d'un millier de bases est parfois codé par un fragment d'ADN qui peut atteindre des dizaines, des centaines, voire un millier de kilopaires de bases. Il est par exemple fréquent qu'un exon très court (quelques paires de bases) se trouve noyé au milieu d'un océan d'introns : la détermination de la séquence d'un fragment d'ADN, même long, peut alors se révéler très insuffisante pour définir le gène qu'il spécifie. En plus des difficultés dues à ce mode de construction modulaire s'ajoute celle de caractériser ce qu'est, au niveau moléculaire, un génome humain. La diversité génétique des populations provient d'une variabilité (polymorphisme) très importante dans les séquences génomiques, y compris chez un même individu lorsqu'on considère la même région d'un chromosome maternel et de sa contrepartie paternelle. Au cours de l'acquisition de la séquence il sera donc très difficile de distinguer ce qui revient aux erreurs expérimentales de ce qui est seulement une variation habituelle (**).
Ce qui nous intéresse, plus que la séquence d'un génome proprement dite, sont les fonctions élémentaires responsables de la vie cellulaire. Il y a environ 250 types de cellules chez un mammifère comme l'Homme, et faire l'inventaire des ARN messagers, matrices où s'élaborent les protéines responsables de la vie cellulaire, alors que beaucoup sont spécifiques du type cellulaire considéré, est une ambition à la fois plus prometteuse, puisqu'elle donnera accès directement au produit des gènes, et plus réaliste, puisque, bien souvent, moins de 10.000 gènes sont exprimés dans un même type cellulaire. L'obtention de la séquence de ces messagers, "étiquettes" est alors un objectif du même ordre que celui de la détermination des séquences de génomes bactériens. Cela implique cependant la mise au point de techniques de tri efficace permettant de séparer les classes de messagers en fonction de leur abondance dans la cellule. A partir de ces données, il sera possible de revenir à chacun des chromosomes et d'y placer les gènes ainsi identifiés. Tout cela met en avant l'intérêt d'une cartographie physique des chromosomes, projet beaucoup plus accessible, au moins dans un premier temps, que le séquençage du génome lui-même. On pourrait obtenir ainsi la localisation précise de gènes identifiés, en particulier de gènes responsables de pathologies diverses. Cet objectif, beaucoup moins ambitieux que le séquençage, est celui qui est désormais fixé pour le programme "Génome Humain". Ses promoteurs espèrent qu'il pourra déboucher sur l'invention de méthodes thérapeutiques nouvelles, compte tenu, on le souhaite, de critères éthiques.
La réflexion scientifique prenant le pas sur la recherche du succès médiatique, il semble essentiel, afin non seulement de développer des techniques permettant le séquençage rapide, mais aussi de reconnaître ce qui est pertinent dans une séquence nucléotidique, de choisir d'abord des organismes modèles. Les génomes mammifères sont très voisins les uns des autres. Afin de diminuer l'importance des difficultés mentionnées plus haut il faut entreprendre l'étude génétique d'un organisme dont le génotype soit aussi homogène que possible. La souris de laboratoire existe sous forme de lignées consanguines, et elle a un génome très voisin du génome humain. De plus l'analyse fonctionnelle de ses gènes pourra se faire par génétique "inverse". Cela consiste à remettre au sein de l'organisme le gène étudié sous forme défectueuse ou altérée, afin d'en comprendre le rôle (et c'est évidemment impossible chez l'Homme). Malgré la lourdeur expérimentale que cela suppose, les constructions de souris transgéniques sont bien adaptées à des études de ce type. Le modèle souris reste cependant un modèle encore inaccessible par sa taille aux techniques actuelles du séquençage de l'ADN.
Il existe d'autres organismes différenciés plus simples chez lesquels l'analyse du génome est possible. C'est le cas de la mouche drosophile, dont la génétique est bien connue depuis les travaux de T.H. Morgan. Son génome est d'environ un vingtième d'un génome mammifère ou 30 fois celui d'une bactérie comme le colibacille. Dans un autre ordre d'idées, comme les stratégies de différenciation des animaux et des végétaux sont très dissemblables, on ne peut se contenter d'explorer les caractères spécifiques aux animaux. Il faut s'intéresser aux plantes. En particulier, tout ce qui relève de la photosynthèse, et des interactions entre le génome des chloroplastes (organites intracellulaires responsables de la fixation du gaz carbonique, et qui donnent leur couleur verte aux plantes) et le noyau est d'un très grand intérêt. La crucifère Arabidopsis thaliana, a un génome qui se réduit à une centaine de mégapaires de bases (Tableau I).
Chez les mammifères, les plantes, ou les insectes un peu élaborés comme la mouche, le plan général, son organisation fine sont conservés, mais le nombre exact des cellules de chaque tissu varie d'un individu à l'autre. Pour cette raison, Sydney Brenner, du Medical Research Council à Cambridge, a proposé il y a une vingtaine d'années de rechercher un modèle de la différenciation où tout le patron de développement serait rigidement fixé. Le nématode Caenorhabditis elegans, ver minuscule des sols, se prête bien à des études de ce type car, à l'état adulte, le nombre et la disposition de ses cellules est fixe, et le schéma temporel de leur différenciation - y compris la mort programmée de certaines d'entre elles - est absolument fixé par ses gènes. Caenorhabditis elegans adulte mâle est composé de 1031 cellules et l'on connaît exactement la filiation de chacune d'entre elles depuis l'oeuf fécondé. Le génome total du ver est formé de 100 mégapaires de bases environ. Un ensemble concerté de travaux menés en Angleterre et aux USA principalement, a permis de réaliser une bibliothèque d'ADN qui recouvre presque tout le génome, et le séquençage a déjà livré la séquence de trois fragments totalisant un peu plus de 100 kb. Le résultat le plus intéressant de ces travaux préliminaires est que la densité des gènes, malgré l'importance des introns, est élevée : on trouve un gène tous les 4 kb.
Mais c'est le cas de la levure de boulanger qui est actuellement exemplaire. André Goffeau, professeur à l'Université Catholique de Louvain, et fonctionnaire de la Commission des Communautés Européennes, réussissait dès 1986, malgré des résistances nombreuses, à convaincre nombre de ses collègues spécialistes de la levure de boulangerie, Saccharomyces cerevisiae, de s'associer pour entreprendre le séquençage du génome de cet organisme simple.
Les motifs de ce choix étaient nombreux. S. cerevisiae est une cellule nucléée, comme le sont les cellules animales et végétales. Son noyau contient 16 chromosomes, et son génome total est d'environ 15 mégapaires de bases. La levure est utilisée en alimentation depuis la plus haute antiquité. C'est l'un des ingrédient principaux de la bière et du pain. Elle est aussi l'un des principaux fleurons du travail de Pasteur, dans sa découverte du rôle essentiel des microbes dans le monde vivant. Par ailleurs, la levure se prête bien à la génétique inverse. Des centaines de laboratoire dans le monde en ont fait, depuis plus de cinquante ans, l'étude génétique poussée. Une association d'une quarantaine de laboratoires européens a conduit à déterminer la totalité de la séquence du chromosome III (315 357 pb), codant 182 gènes, début 1992.
Cette étude, soutenue par la communauté européenne, a d'abord été financée par le programme BAP, puis par le programme BRIDGE, et un projet de quatre années, financé par le programme BIOTECH, devrait conduire ces mêmes laboratoires, associés à quelques autres, à séquencer plus du tiers du génome d'ici 1995 (***). Le programme fut organisé par André Goffeau de la façon suivante. Un coordinateur, Stephen Oliver (professeur à l'université de Manchester), devait construire une banque de fragments de l'ADN de ce chromosome et les distribuer aux différents laboratoires au fur et à mesure des besoins. Le contrôle de qualité était effectué grâce aux recouvrements qui existaient entre les différents fragments séquencés par différents laboratoires, d'une part, et d'autre part grâce au séquençage, en double, par des laboratoires indépendants, de régions semblables mais provenant de clone différents de la banque. Enfin un coordinateur pour l'analyse fonctionnelle des gènes, Piotr Slonimski du centre de génétique moléculaire de Gif sur Yvette, fut désigné dès qu'il apparut que des questions importantes se posaient pour identifier la fonction des gènes séquencés.
D'ores et déjà, en effet, deux résultats très intéressants sont apparus. D'une part, le génome est très compact (il y a très peu d'ADN redondant ou, en apparence, non signifiant). D'autre part, plus de la moitié des gènes correspondent à des protéines qui ne ressemblent à rien de connu (dans les banques de données, qui contiennent environ 100 Mpb d'ADN provenant d'organismes variés). Ces gènes spécifiant des protéines énigmatiques, ont été appelés EEC gènes (acronyme de elusive, expressed, conspicuous genes) par P. Slonimski au cours d'une réunion de concertation internationale à Elounda en Crète en mai 1991, pour rappeler le rôle pionnier de l'Europe dans leur découverte. Ils étaient totalement inattendus. On sait cependant à la suite des travaux de K. Isono et de A. Yoshikawa de l'Université de Kobe qu'ils sont exprimés, et que les protéines dont ils dirigent la synthèse doivent donc avoir un sens. Que signifient-ils ? Comment avoir accès à leur fonction ? C'est là un des défis que ces premiers résultats jettent à la génétique. Et c'est d'autant plus difficile qu'au premier abord, dans la majorité des cas, l'inactivation de l'un ou l'autre de ces gènes n'est suivie d'aucun effet apparent ! Il est bien certain que les conditions de laboratoire sont très différentes de ce qui se passe dans la nature, mais comment mettre au point toutes les conditions de culture de la levure qu'il faudrait explorer ? On peut avoir une idée de la difficulté du problème en sachant que l'inactivation d'un gène, correspondant à une très longue protéine, analysée dans le laboratoire de P. Slonimski ne montra un effet que lorsque le milieu de croissance contient de l'acide acétique, à un pH inférieur à 4,5… Il faudra donc combiner toutes sortes de connaissances et d'observations pour tenter de mettre au jour toutes ces nouvelles fonctions. Un moyen de le faire sera de recourir à toutes les techniques possible permettant de classer les gènes, par leur structure ou par leur expression, en familles ayant un comportement homogène. On peut espérer ainsi constituer des familles au sein desquelles il y aura des gènes connus, et diriger alors l'exploration à partir de ce premier pan de connaissance.
Voilà donc beaucoup de raisons pour poursuivre une étude si bien commencée. Déjà le projet structuré de la même manière autour des chromosomes II et XI est bien avancé, et A. Goffeau pense que leur séquence complète sera sans doute accessible dès la fin de l'année 1993. La moitié de la séquence du génome pourrait bien être connue fin 1995 (****). La situation du projet de séquençage, hors de l'Europe, est un peu plus complexe. En effet il semble qu'il n'ait pas été pour l'instant possible d'obtenir un consensus avec les laboratoires américains intéressés par ce projet. Cependant, au Canada, en Australie et au Japon des ensembles concertés vont se consacrer au séquençage de chromosomes individuels. Il est alors probable que devant le succès de ces approches les laboratoires américains vont se soumettre à la règle commune et abandonner d'inutiles prétentions de priorité. Il convient ici de remarquer que le programme "Human Genome" qui, sous la direction de Jim Watson, refusait de considérer comme pertinents les projets autres, est désormais dirigé par Francis Collins, qui pourrait avoir une philosophie bien différente.
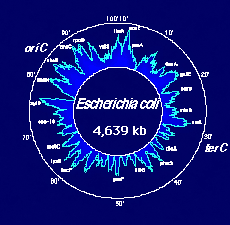
En raison de l'extrême compacité de leur génome (tout semble y être signifiant), les bactéries sont les organismes qui semblent se prêter le mieux à l'étude, et pourtant elles n'ont été impliquées réellement dans les projets de séquençage que récemment. Nous disposons d'informations génétiques nombreuses à leur sujet. Par ailleurs, elles ont un intérêt évident pour l'environnement, l'industrie ou la médecine. Enfin, elles peuvent être manipulées aisément. Le parangon bactérien reste le colibacille, Escherichia coli, dont le chromosome unique est formé de 5020 kpb. Plus de 1100 loci génétiques ont déjà été identifiés chez cette bactérie et de très nombreux gènes en ont été séquencés (1950 kpb, mi-1992). Il lui reste malheureusement un handicap spécifique : bien qu'elle soit possible, la génétique inverse ne s'y réalise qu'assez laborieusement. Par ailleurs il semble bien qu'il existe des rivalités, plutôt qu'un effort concerté devant conduire au séquençage complet de son génome. Un groupe de laboratoires japonais va cependant séquencer environ 600 kb en trois ans, autour du gène choisi comme origine de la carte génétique : les premiers 100 kb ont été envoyés à la banque de données japonaise fin juin 1992. Et le laboratoire de Blattner aux Etats Unis a publié en août de la même année une zone contigue de 90 kbp, dans une région dèjà bien connue…
Quels autres efforts sont possibles ? Il faut ici à nouveau mêler la politique scientifique à la recherche, afin de comprendre comment s'édifient ces projets. Convaincu de l'intérêt scientifique immense de la connaissance totale de génomes, au cours d'une réunion organisée par la Société Française de Microbiologie, début 1987, je proposai que se mette en place le séquençage d'un génome bactérien. A cette époque il était couramment admis que le séquençage du génome du colibacille était presque terminé (ce bruit était répandu par Blattner ou Church aux USA, ou encore par Kohara et Isono au Japon). Il était donc intéressant d'envisager le séquençage d'un autre organisme, en utilisant le savoir-faire correspondant à E. coli. La bactérie la mieux connue après E. coli était Bacillus subtilis, bactérie formant des spores, chez laquelle la génétique inverse se réalise très facilement. Son génome (4200 kpb) est moins bien connu, mais plus de 700 gènes ont été localisés. Plus de 400 kpb d'ADN déjà séquencés existent dans les banques de données et quelques ensembles concertés de laboratoires aux Etats-Unis, au Japon et en Europe collaborent depuis de nombreuses années à son étude. Cependant les contacts que je pris à cette époque avec les laboratoires français spécialistes de cet organisme furent négatifs.
Au cours de l'été 1987 un événement favorable vint fort heureusement inciter de nombreux chercheurs à s'impliquer dans un projet international de séquençage du génome de B. subtilis. Au cours de la réunion annuelle des chercheurs intéressés par cet organisme Jim Hoch (Scripps Clinic à La Jolla en Californie) proposa en effet la mise en place d'un groupe, mi-européen, mi-américain, qui se partagerait l'effort de séquençage. Raymond Dedonder, alors directeur de l'Institut Pasteur, et spécialiste de B. subtilis en revint persuadé, et il décida de tenter de créer le projet européen. Un accord fut obtenu rapidement entre six laboratoires, qui, dans une étape initiale, devaient coopérer avec cinq groupes américains. Le projet américain soumis au National Institute of Health ne fut pas accepté, alors que le projet européen, coordonné par R. Dedonder, fut financé par le programme SCIENCE de la CEE. Dans un premier temps il était décidé que seraient constituées des banques du génome de la souche 168 et qu'un premier effort de séquençage devait tenter de démontrer la faisabilité du projet. Ce programme devait coordonner les efforts de cinq centres européens : deux en France, à l’Institut Pasteur et à l'INRA de Jouy en Josas, un en Irlande un en Angleterre et un en Italie.
En parallèle les groupes américains réunis autour de Jim Hoch tentaient à plusieurs reprises, mais sans succès, de voir leur programme soutenu par le NIH ou le DOE. La situation actuelle est que nous ne pouvons pas, malheureusement compter à très brève échéance sur la collaboration des américains. La situation est fort différente avec le Japon. Au cours d'une réunion, en septembre 1990, destinée à faire le point sur les premiers résultats de ces efforts concertés, H. Yoshikawa signala que le Japon était désireux de s'associer au programme. Un ensemble coordonné de six laboratoires japonais est donc maintenant associé à l'effort européen. L'effort du Japon, sous la responsabilité directe de H. Yoshikawa, est partie intégrante de l'effort général du Japon en matière de séquençage des grands génomes, coordonné par K. Matsubara.
À la mi-1992, six mois avant le lancement effectif du programme CEE BIOTECH, les résultats sont déjà fort prometteurs. Trois banques de gènes ont été constituées, conformément aux prévisions. De plus, à l'Institut Pasteur, P. Glaser a mis au point une stratégie efficace de "marche sur le chromosome", qui permet de déterminer la séquence de longs fragments contigus. Il est parvenu à organiser le séquençage de plus de 100 kb, en distribuant les projets sous la forme de fragments de 8 à 20 kb à des chercheurs ou des ingénieurs qui devenaient responsables pour des périodes de quatre à six mois le plus souvent d'un projet complet, depuis le clonage initial jusqu'au séquençage et la génétique inverse. Le travail, de puis l'isolement initial des fragments considérés, jusqu'à l'étude par génétique inverse des gènes les plus intéressants, prend environ six mois. Ces "pochettes surprises" ont livré jusqu'à présent des résultats étonnants. En dehors de gènes déjà connus, 92 séquences codantes ont été trouvées, parmi lesquelles 35 seulement ressemblaient à des gènes connus dans d'autres organismes. Parmi celles-ci nous avons identifié des gènes de biosynthèse d'antibiotiques, de dégradation de sucres ou d'acides aminés, et de systèmes respiratoires. Dans les autres laboratoires (essentiellement à Jouy en Josas, où le laboratoire de D. Ehrlich a suivi une stratégie différente, tendant à séquencer des fragments beaucoup plus longs, isolés dans des chromosomes artificiels de levure et à Pavie, dans le laboratoire de A. Galizzi) des résultats du même type, portant sur des fragments contigus de l'ordre de 30 kpb ont été trouvés. Au total plus de 160 kbp sont connus.
Comme on peut le voir il s'agit là d'une véritable mine génétique à exploiter. Le résultat sans doute le plus surprenant et le plus intéressant est le grand nombre des gènes qui ne ressemblent à rien de connu. Comme pour la levure, le séquençage "au hasard" de fragments du génome de B. subtilis a révélé l'existence d'un très grand nombre de gènes (près des deux tiers de ceux qui ont été identifiés) dont le produit ne s'apparente à aucun gène connu. Il s'agit d'une observation très remarquable, qui démontre que les approches génétiques classiques ont laissé de côté un pan immense de notre connaissance des organismes, dont nous n'aurions même pas soupçonné l'existence si nous n'avions commencé à mettre en place des projets de séquençage de grands génomes. Cela seul suffirait donc à justifier de tels projets.
Ce résultat est très paradoxal, et met en question notre savoir en génétique. En effet, si l'on isole dans la nature un individu quelconque différant du type "normal" de son espèce (un mutant), d'une espèce quelconque (de la bactérie à l'homme), et qu'on caractérise par séquençage le produit du gène concerné, on trouve dans plus de 9 cas sur 10 qu'il ressemble à quelque chose de connu. Que signifient alors ces gènes inconnus ? Pour le comprendre il conviendra de créer des méthodes nouvelles permettant d'analyser la fonction de ces protéines nouvelles, car il va de soi que leur existence même fait penser que des gènes semblabless, et sans doute beaucoup plus nombreux se trouveront chez les cellules supérieures, alors que nous n'aurons aucun moyen d'accès rapide à leur fonction. Les modèles bactériens se révéleront alors sans doute indispensables. Ainsi, au contraire de ce qui a été naguère dit aux Etats Unis, les modèles microbiens ne doivent pas suivre le programme du génome humain, mais plutôt l'accompagner, et même le précéder, pour lui permettre de prendre tout son sens.
En parallèle, l'effort informatique devra à terme être aussi important que l'effort de séquençage. En effet, contrairement à une opinion superficielle, malheureusement très répandue, la nécessité d'une véritable recherche en informatique, découlant tout naturellement des projets de séquençage, n'est aujourd’hui que très peu prise en compte dans les modes d’analyse des génomes dont on dispose. On ne possède en fait, le plus souvent, qu’une table de traduction, utilisant le code génétique, et de banques de données permettant de comparer les nouvelles acquisitions à ce qu’on connaît déjà. Cela ne serait pas si mal si l’on connaissait presque tout, mais ce n’est en aucune manière un moyen d’innovation. D’une façon générale on pense qu’il suffit d’obtenir la séquence d’un gène, et de son environnement immédiat pour savoir la plupart de ce qu’on souhaite à son propos. Si cela est vrai dans un premier temps - et ne nécessite qu’une infrastructure informatique légère permettant d’avoir un accès aisé à tout ce qui est contenu dans les banques de données - c’est infiniment loin de ce qu’est le dessein ultime des projets de séquençage : comprendre le fonctionnement d’un génome. On pourrait croire, par exemple, que découvrir qu’une protéine ressemble à une enzyme connue suffit à déterminer sa fonction, mais que dire de cette naïveté lorsqu’on sait, par exemple, qu’une lactate deshydrogénase peut être une protéine du cristallin de l’œil, dont la fonction principale est d'assurer la transparence ? En fait on ne sait bien retrouver que ce que l’on connaît déjà. Que faire lorsqu’on est en face d’une séquence qui ne ressemble à rien de connu ? Il ne s’agit nullement là d’une hypothèse d’école, on l'a vu, car le séquençage du chromosome III de la levure, de longs fragments du génome de B. subtilis, et du début d’un grand nombre d’étiquettes de lignées cellulaires humaines montre que c’est en réalité la situation générale. Nous nous trouvons là en face d’une situation semblable à celle de l’archéologue qui découvre une langue inconnue, à partir de très nombreux textes. Et ce qu’il nous faut donc faire, c’est créer les moyens de construire une pierre de Rosette pour les comprendre.
L'informatique intervient dans ces projets à trois niveaux principaux : lors de l'acquisition des données, lors de leur exploitation et lors de leur gestion. A chaque niveau correspondent des questions spécifiques à la fois pour les biologistes et pour les informaticiens. Le foisonnement des données, la complexité et la variété des connaissances que l'on a à leur propos sont autant de défis aux chercheurs de chacune des disciplines. Les projets de séquençage sont donc par essence interdisciplinaires, en ce sens qu'il ne peut être envisagé à aucun moment qu'une des communautés soit au service de l'autre (les biologistes fournissant des données ou des questions, ou les informaticiens fournissant des programmes). Leur succès démontrerait qu'il est possible d'ouvrir de nouveaux champs de recherche à l'interface entre deux disciplines, et où l'association d'expertises, non hiérarchisée, conduit à la découverte. Le point sans doute central de cette interaction est que le retour à l'expérience, au travers de la construction d'objets biologiques artificiels (gènes et protéines) sera un moyen particulièrement puissant de validation à la fois des créations informatiques, et des créations biologiques. Il existe en science très peu de situations ayant le degré de généralité que permet la manipulation in vitro des gènes. Et l'on peut à juste titre penser qu'aux approches habituelles de la biologie, expérimentation in vivo et in vitro, s'ajoutera la dimension nouvelle de l'expérimentation "in silico".
Un domaine de l'informatique directement concerné est bien sûr la recherche en intelligence artificielle et en apprentissage : que ce soient les questions de reconnaissance des formes, ou d'analyse des textes,. Mais cela n'exclut pas des domaines dont la thématique est plus classique, ou plus ancienne : combinatoire, algorithmique, traitement du signal, bases de données et, pour l'étude des relations entre objets, théorie des graphes, par exemple. Cela n'exclut pas non plus des domaines spécifiques plus à la mode, comme la conception ou l'utilisation des réseaux neuromimétiques. Il faut noter par ailleurs que, dans la mesure où les données génétiques sont le résultat d'une histoire (l'évolution des espèces) leur accumulation même très importante ne permettra jamais de les comparer à ce qu'aurait engendré le hasard. Il s'en suit que les méthodes de classification des objets sont d'une importance cruciale, et doivent être considérées en priorité dans beaucoup de cas. En effet, une mauvaise agrégation des objets initiaux conduit, même avec des méthodes très performantes à des résultats décevants. Il s'agit en effet d'intégrer un ensemble de connaissances disparates en vue de la prédiction des structures et des fonctions. La cohérence de l'évolution qui a permis un ensemble immense d'essais et d'erreurs successifs pour arriver à un certain degré de perfection dans l'activité des enzymes représente une information beaucoup plus riche que celle qu'on peut obtenir simplement par mutagénèse localisée. Malheureusement, cette information est difficilement accessible. On peut cependant tenter d'y avoir accès au moyen de techniques d'apprentissage sur des lots d'exemples où l'on essaie de découvrir par assimilation ou par discrimination des propriétés d'un ensemble bien constitué.
Cela suppose une grande intégration des approches informatiques, de façon à pouvoir mettre ensemble les diverses connaissances biologiques, et les connaissances qui peuvent dériver d’une analyse plus mathématique ou physique des séquences de nucléotides obtenues. Cette intégration doit se faire dès l’acquisition des données, car il y a dans le fait même d’obtenir la séquence d’un fragment d’ADN, toute une connaissance biologique. Les difficultés de clonage, les erreurs de séquençage, le polymorphisme, les anomalies de migration sur gels peuvent tous donner des informations précieuses sur la nature du fragment étudié (on a pu noter, par exemple, des anomalies de migration dans les régions qui peuvent former tige et boucles à la fin des transcrits…). Il faut ensuite analyser les données, les comparer à ce qui est connu, les intégrer dans les différents projets, puis les gérer efficacement. Il faut enfin les communiquer à la communauté des chercheurs sous une forme aussi riche que possible, de façon à faire profiter toute la communauté de l’apport d’un travail gigantesque. Par exemple les données générales rassemblées dans le cas de E. coli (dont le séquençage est principalement le fait de la collaboration implicite de tous les laboratoires du monde travaillant sur l'organisme, et non le fait de laboratoires spécialisés dans le séquençage des grands génomes, du moins pour l'instant) sont disponibles dans plusieurs banques de données. Celles-ci nous ont servi à établir un protocole de fabrication de bases de données spécialisées, c'est à dire non plus seulement de recueil de données telles qu'elles apparaissent dans la littérature, mais de données qui évoluent au cours du temps, au fur et à mesure de l'incorporation de nouvelles connaissances, pour E. coli et B. subtilis.
On n'a pu, jusqu'à présent, faute de connaître la séquence de génomes entiers, explorer leurs caractéristiques globales. Cela sera particulièrement efficace dès qu'il sera possible de comparer des fonctions identiques chez des organismes phylogénétiquement éloignés. Mais le nombre des gènes présents dans un même organisme et descendant d'un ancêtre commun est déjà assez grand pour permettre d'identifier certaines règles essentielles, à partir d'un seul génome. C'est le cas par exemple d'une découverte que C. Médigue, A. Hénaut et moi-même avons faite dans le cas du colibacille. En analysant, au moyen de techniques de taxinomie statistique, la nature des gènes connus chez E. coli nous avons découvert qu'il était possible de les classer en trois familles. Deux de ces familles avaient déjà été identifiées, il y a longtemps par R. Grantham et C. Gautier, de l'Université de Lyon. Mais la troisième famille était surprenante. Elle semblait rassembler, sous la contrainte d'une pression de sélection s'exerçant surtout sur l'ADN lui-même (et non sur les gènes qu'il spécifie), des gènes impliqués dans le transfert de fragments d'ADN entre cellules différentes. On trouvait en effet dans cette classe les gènes de la "sexualité" bactérienne, des gènes apparentés à ceux des virus tempérés, des plasmides, ou encore des transposons, qui se promènent d'un endroit à l'autre des chromosomes. Mais le plus remarquable peut être était que dans cette famille se trouvent les gènes connus pur être "anti-mutateurs", c'est à dire capable de corriger les erreurs qui se produisent immanquablement au cours de la réplication de l'ADN. Cette observation conduit à l'hypothèse que dans la nature beaucoup de bactéries ne constituent pas à proprement parler des espèces, mais sont des individus à la descendance hautement variable, qui se fixeraient en espèce, lorsque l'environnement s'y prête, par transfert horizontal de gènes anti-mutateurs. Si cela est exact — et des données récentes caractérisant des bactéries isolées vont dans ce sens — le transfert génétique dans la nature serait un phénomène universellement et constamment à l'œuvre ! Voilà une donnée importante qui devrait être prise en compte lorsqu'on modifie la flore des sols par exemple.
Cet article écrit par Antoine Danchin, le 22 août 1992 est le
manuscrit d'un article paru dans La Recherche © 1993
Le séquençage des petits génomes. Vers la description complète d'un organisme vivant
La Recherche (1993) 251: 222-232
Version espagnole: La Secuenciacion de pequeños genomas. "Hacia la description completa de un organismo vivo"
Mundo Cientifico (1993) 134: 376-386
On trouvera la traduction en anglais de ma vision présentée en 2000 du programme génome humain
A Danchin
Une histoire intense, presque violente: l'histoire du programme génome humain [A rattling good history: the story of the Human Genome Project]
La Recherche (2000) 332
On mesure systématiquement la longueur des séquences génomiques en kilobases (kb), mégabases (Mb) et même gigabases (Gb) par multiples de 1000
(*) On évaluait à l'époque le nombre des gènes humains à 100000 au moins. Les travaux de séquençage du génome ne permirent d'en identifier qu'un peu plus de 30000, ce qui a paru paradoxal à la pensée superficielle qui confond le quantitatif et le qualitatif: on sait bien que la standardisation des composants permet néanmoins la construction d'une infinie variété d'objets.
(**) À l'époque il était très difficile de séquencer plus de 50 kb par personne et par an; au surplus le taux d'erreurs de séquençage était élevé. A partir de 2000 les progrès techniques ont été tels et le taux d'erreur si bas qu'il devenait possible d'identifier le polymorphisme individuel, et de s'en servir pour étudier les mouvements de populations, aussi bien que l'origine de maladies génétiques diverses. Ces SNPs (Single Nucleotide Polymorphism) sont à la base d'un énorme projet, HAPMAP qui tend à identifier l'ensemble du polymorphisme humain.
Il s'agit là vraiment de génétique inverse, et non de ce que par abus de langage certains ont appelé du même nom, et qui consiste, connaissant le code génétique et le séquence d'un gène, à induire la fonction de son produit, par comparaison avec ce qui est déjà connu.
L. Pasteur (1876) Etudes sur la bière. Gauthier-Villars, Paris
BAP : Biotechnology Action Program ; BRIDGE: Biotechnologie pour l’Innovation, le Développement et la Croissance en Europe; BIOTECH : Biotechnologie
(****) voir plus haut
National Institutes of Health ; DoE : Department of Energy