Amongst the
questions asked by the Oracle of Delphi, Pythia, was a fundamental
question directly related to the nature of the artefacts produced and
used by living organisms – an enigma, as the Oracle’s
questions always were. If we consider a boat made of planks of wood,
carefully fitted together, we may well ask, what is it that makes the
boat a boat? This question is more than just a mind game, as is clear
from the fact that as time passes, some of the planks begin to rot
and have to be replaced. There comes a time when not one of the original
planks is left.
The boat still looks like the original one, yet
in material terms it has changed. Is it still the same boat? The owner
would certainly say yes, this is my boat. Yet none of the material it
was originally built from is still there. If we were to analyse the components
of the boat, the planks, we would not learn very much. We can see this
if we take the boat to pieces: it is reduced to a pile of planks – but they are not the same ones as at
the beginning! The physical nature of these objects plays some role of course
– a boat made from planks of oak is different from a boat made from planks
of pine – but this is fairly incidental. (It is very important to remember
this when we think about the possibility of life existing elsewhere in the universe
– there is absolutely no reason why it should be made of the same molecules
as life on Earth.) What is important about the material of the planks, apart
from their relative stability over time, is the fact that it allows them to
be shaped, so that they relate to each other in a certain way. The boat is
not the material it is made from, but something else, much more interesting,
which organizes the material of the planks: the boat is the relationship between
the planks. Similarly, the study of life should never be restricted to objects,
but must look into their relationships. This is why a genome cannot be regarded
as simply a collection of genes. It is much more than that.
This starts a cycle where biological knowledge is integrated with the genome text analysis, permitting the scientist to make predictions, which must be tested in vivo by reverse genetics (where altered genes are made to replace their normal parent in situ) and in vitro by characterizing the gene products and their interactions.

A first
prediction: there is a map of the cell in the chromosome
There is now no doubt that genes are not distributed randomly
in the chromosome of a bacterium such as E. coli. And this is visibly
linked to the function of the proteins specified by the genes, and by the
architecture of the cell. This observation is, at first sight, a mystery:
how can one understand, indeed, the link which must exist between a symbolic
text, the gene text, its products and an architecture? For a correspondence
to exist there must exist a physical link between these different aspects
of Reality. There must exist, somewhere between the gene, its product and
the place where it must be located in the cell, some targeting process. And,
if we are to stay with the simplicity of Ockham's razor — not multiply
the hypotheses — we must look for simple physico-chemical principles.
Let us now proceed somewhat as did the presocratic philosophers when they
investigated the necessary constraints operating on the world — with
the obvious risk to be far too general and too unprecise, but with the hope
for trying new paths for our investigation. Let us reason using symmetry (not
in shape of course, but in the nature of physical laws) as a basic principle.
- A few principles of geometry and physics: helices and hexagons
Let us study the becoming of the product of a single gene,
a protein, synthesized on a ribosome. It may face two situations. Either it
does not interact with itself, and the absence of specific constraints (this
represents the true meaning of entropy) will lead it equally well in all directions,
in all locations in the cell. Or it has some affinity for itself (the case
for repulsion, theoretically possible, is rare with biological objects, at
least at the molecular scale, if not at the cellular scale). Some region of
its surface, A, will interact with region B of a second molecule of the same
protein. This makes a dimer. But this dimer possesses on the first subunit
a free B region, while the second subunit has an A free region. This is of
course the most general situation, but there are special situation where A
and B contact each other, creating a dimer with a symmetry similar (in three
dimensions!) to that of the Yin and Yang figure. In the general case however,
while the ribosome synthesizes a third instance of the protein, this latter
one will have the same tendency as the formers, to associate to the complex
they form. But this association is not random: it must occur mainly at the
particular regions of the proteins making a region A, face a region B, in
the proper orientation. This process will continue as more proteins are synthesized.
In the most general case this yields an helix structure. This simple reasoning
shows that the helix is the first among the biological forms. It is therefore
the most frequent and the most trivial one. Until now, nothing very surprising,
but the acknowledgement of the existence of an essential form, dissymetrical
by construction, at the root of all living forms (and we find here the word
of Pasteur: "Dissymmetry is life!").
Another
remarkable consequence of this property is that helicoidal forms permit
the cell to make length measurements with an exquisite precision! It
is what happens during the formation of the tail of some viruses (an
appendix which serves them to inject their genome into the host cell).
The trick is to uses a couple of helices with a different pitch, one
helix making a tube into which the second one fits. These pitches of
course have to be commensurable (but this is always true at some length,
because of the thermal fluctuations underwent by all biological structures).
Beginning at the same point, the helixes begin to part from each other.
But after a certain number of turns the extremities of both helices are
once again in the vicinity of each other on the same radius of the base
circle. This creates a context where the construction can be terminated.
Subsequently, a process continuing the construction of the virus (the
construction of its head), triggers the depolymerisation of the internal
scaffolding helix, made of identical subunits, which are released in
the medium, leaving a hollow tail having a well-defined length. This
process, which uses the adequation of the folding of two helices is certainly
very general. It derives directly from the use of the ubiquitous helical
form, transforming the constraints of necessity into an elaborate means
to create new formal properties, such as that of measuring length. In
the same way, time may also be measured by several kinds of processes,
usually quite simple. Because nucleic acids (RNAs and DNA) make also
helices, they constrain very strongly the systems which interact with
them, allowing, once again, for proper positioning, and this may be the
mechanism which allows the enzyme (telomerase) that take charge of the
extremities of the chromosomes when they are linear, the telomeres, to
restore their length when they have been shortened by a succession of
replication events.
Evolution is exploration, and, as proteins aggregate into
helicoïdal structures they will explore all kinds of pitches, with many
different base circles, leaving in their inside a more or less wide hole,
as genes vary by mutations. Among the explored pitches is the flat pitch (that
is, in which all subunits stay in a plane instead of building up an helix).
This leads therefore to cyclic structures. The geometrical properties of all
these structures have been studied since Antiquity. They comprise, in particular,
the regular polyedra dear to Plato, which he describes in Timeos. And, as
a matter of fact, such polymers do exist in many biological structures. They
form, for example, many viral capsides. Now, a construction of this type (an
icosaedral virus, for instance), possesses a simple property, that one identifies
easily following the line of reasoning which was followed until now (this
was done considering single subunits): if it does not interact with something
particular, it will naturally tend to explore the whole cell. At some point
it will, therefore, meet the cell membrane. This is what will allow it to
get out of the cell.
The largest increase in entropy of a molecular complex in
water is when its surface / volume ratio is the highest. This is the case
especially of plane layers, and this is minimised in spherical structures.
As a consequence, when a plane meets another one, it can loose a layer of
water molecules and stick there, piling up. Among all these structures there
exist one which has a remarkable property, it is the hexagone. Indeed, regular
hexagones will necessarily form a plane paving if they interact, a structure
similar to that discovered by bees in their nests. Many geometrical structures
can lead to planar structures, but regular hexagons have only two ways to
interact: either they pile upon each other, forming tubes, or they form planes.
Indeed many membrane structures are made of planar layers, and in particular
of pieces of hexagonal paving. Let us imagine what happens to a piece of hexagonal
plane, made of subunits being synthesized in a ribosome, associated to the
corresponding messenger RNA. This fragment will spontaneously move away from
its biosynthesis place, either following the trend of the fluid movements
inside the cell, or moving along an electrostatic gradient, or any other form
of diffusive movement. Roughly speaking, it will tend to escape from the place
where it is. This means that it will move until it reaches some obstacle.
And the first obstacle it will meet has also the property to be, locally,
a plane, therefore preadapted to interact with a piece of plane paving, this
is one of the cell's membrane structures! Will this piece of plane bounce
back, and go back inside the cell? In most cases, certainly not. In water,
a piece of plane imposes that the water molecules in its neighborhood have
only a limited number of available positions and states. Its presence, amid
the water solution, is therefore extremely constraining in terms of entropy.
If, in the course of its diffusion in the medium, it meets with a planar surface,
the water molecules present on the layer at the interface will be eliminated
into the solution, where they can meet a much larger number of positions and
states. The piling up of such structures, at the moment when they loose one
water molecule layer at their surface will therefore be extremely favored
by the second principle of thermodynamics.
One may therefore expect that all hexagonal structures, or
any other more complex structure making plane pavings, will be rapidly piling
up under the cell's membrane. This will happen without any special requirement
for specific electrostatic charges (there can exist however a small electrostatic
effect, which will direct more rapidly the fragment of plane toward the surface),
or specific interactions with membrane lipids, to account for this original
architectural property. What makes the principle of the interaction is simply
the fact that planes pile up easily, simply because they are plane. As a matter
of fact, when compared to other genometrical structures, the plane provides
the largest ratio, by far, of its surface as compared to the volume of the
object it constitutes, and it leads to the largest entropy contribution at
the moment when it piles onto another plane. One can easily conceive, then,
that this very simple physical principle — which uses the natural propensity
of things, the shi of the Chinese, to go towards the direction of an increase
of entropy — may have the property to act as a guiding principle in the
construction of the cell's architecture. It is still too early to be sure
of this prediction. But the first results which we obtained when we studied
the structure of the E. coli genome suggest that the distribution of
the genes which code for proteins making hexagonal pavings appears to display
some regularity along the chromosome.
To conclude and summarize: a distinctive feature of living
organisms is the ubiquitous presence of membrane structures. In fact a general
"strategy" of evolution has been either to compartmentalize the
cell with a single — albeit sometimes very complex — envelope, made
of a lipid bilayer, or to multiply membranes and skins. Once again, this structure
corresponds a posteriori to an efficient way to use the natural tendency
of things to increase their entropy for the following reason. Liquid water
is a highly organised fluid, which has as a built in principle the natural
tendency for water molecules to occupy as many energy and spatial states as
possible. This leads to systematic demixing of those molecules which are in
contact with water molecules, unless they provide energy favoured interactions.
As a consequence, the increase in entropy is the driving force for the construction
of many biological structures: this physical parameter is at the root of the
universal formation of helices, it drives the folding of proteins and the
formation of viral capsids, it organises membranes into bilayers and creates
higher complex biological structures. It already seems certain that as more
and more new genome texts will be deciphered, corresponding to cells with
a variety of architectures (or submitted to strong physical constraints such
as cold conditions, for example), the concrete processes used in the construction
of their parts, associated to the mechanisms of evolution which led to the
present state, will be better and better understood. In particular, while
it is still difficult at the turn of the century to understand the complicated
structure of eucaryotic cells (characterized by the multiplication of membrane
structures: nucleus, endoplasmic reticulum, vacuoles, a variety of organelles,...)
we shall understand progressively how this organization relies to that of
the genome, and in particular to the split nature of the genes, which are
fragmented into introns and exons. One may already think that the piling up
of planar structures play there an important role: if the layers of the endoplasmic
reticulum are synthesized from centers located in the vicinity of the nucleus,
then one may see this synthesis as putting into play a kind of rolling carpet,
which carries along in the cell the translation machinery associated with
the messenger RNAs and their products.
- The order of the genes is not random in the genome
If these physical constraints operate, they must be visible
somehow in the genome text. Indeed, they indicate a compartmentalization of
the gene products, which have therefore to be synthesized in a coordinate
fashion both in time and in space. For genes expressed at a very low level,
or rarely, this is probably not too important (we have however uncovered an
original distribution
of essential genes, even when expressed at a low level), since the diffusion
time may be enough to allow the gene product to explore many of the situations,
but this is not so for genes expressed at a high level, or frequently. Then
there must be some kind of correlation between the neighborhood of the genes
in the chromosome, and that of their products in the cell. We have observed
that there exists a strong bias in the base composition of the leading and
lagging DNA strand in many bacterial genomes. This is true despite the fact
that the way DNA is managed (as witnessed by the variation in the
number of repeats present in genomes) varies immensely from a species
to another one. This indicates that, in contrast to what is often stated,
genomes are rather rigid entities, which do not allow much change in
gene order (unless the change are symmetrical with respect to the origins
of replication). There is indeed lateral gene transfer — and our
studies were the first to demonstrate this convincingly — but gene
transfer does not occur at random. There are hot spots where foreign DNA can
be placed (in particular this seems to be the case of the terminus of replication
in bacteria).
If a DNA molecule the length of the E. coli genome
were coiled randomly, standard polymer theory tells us that it would fit a
sphere with a diameter of ca 10 micrometer at physiological salt concentration:
ten times more than the diameter of the cell. Superordered structures of DNA
must therefore account for the DNA packaging in the cell. They include supercoiling,
organisation into domains, and attachment to specific sites. Are these physical
constraints reflected in the genome sequence? Preliminary studies with the
yeast genome suggested indeed that such structures exist in this organism.
We can notice here that the ability to pack DNA into a small compartment is
a strong selection pressure explaining the existence of a structure such as
a nucleus: this limits considerably the number of available states of the
molecule and allow organisation of its behavior. This means that the number
of degrees of freedom offered to DNA increases when the compartment grows
(the cell or the nucleus). As a consequence, there is a spontaneous (entropy
driven!) tendency of replicating DNA, to occupy the new space offered by cell
growth, creating a natural process for DNA segregation into the daughter cells.
The main folding problem of long polymers such as DNA or RNA
is indeed that they have a great many number of possible states. If they were
free to diffuse, this would be incompatible with any organisation of the cell
architecture. In fact, a set of freely moving long polymers would rapidly
tangle into an unsortable bulk of knotted structures, even if they diffused
through an organised lattice (such as the ribosome lattice). There is a way
out, however. Anchoring points provide a very efficient way to drastically
lower the number of states available for polymer conformations. A single anchoring
point, as is assumed in the general models of transcription, would already
strongly restrict the number of explored states. As seen with what happens
with uncombed long hair, this might not be sufficient to reduce enough the
number of states that transcripts would still be able explore, but this would
limit the formation of knots. It is well established that two anchoring points
instead of only one would limit drastically the exploration of possible states
and reduce it to a manageable number. How could this be achieved in the cell?
A first answer stems from the observation of electron microscopy
images of the translating machinery. It is observed that, along of a messenger
RNA molecule, the ribosomes are spread in a remarkably regular order.
But the physical organization of the cell has no reason to follow the
genetic information flow, which goes from DNA to RNA to protein. This
is a purely conceptual view which, although it is unfortunately spread
into the vast majority of text books, has the defect of being utterly
unrealistic. In contrast, it it most likely that the ribosomes, organised
as a slowly moving lattice, control the nature of gene expression. Nascent
RNA coming off DNA is pulled by a first ribosome which scans for the translation
initiation region (ribosome binding site and initiation codon) and begins
to translate, then by the next one, as in a wire-drawing machine. Considering
that most of the cell's inertia and that the major part of the cell's
energy is in the translation machinery (it is energy costly to synthesize
mRNA, but one mRNA molecule is translated at least twenty times, and energy
is used for loading aminoacids on tRNA and elongating the polypeptide
chain in the ribosome, with concomitant displacement of the mRNA thread),
it is the structure of the ribosome network that organises the mechanics
of gene expression. Translation / transcription coupling makes DNA move
and brings at its surface new genes ready for transcription. The messenger
RNA passes from a ribosome to the next one, controlling synthesis of the
protein it specifies at each ribosome. In passing, we can note that this
process ensures that the distribution of proteins in the cell does not
from a three dimension diffusion process (which would be very slow) but
by the simple linear diffusion of the messenger RNA through the ribosome
lattice. Finally, as soon as an appropriate signal reaches the ribosome
at the same time as the translated messenger, this triggers degradation
of the mRNA from its 5'-end using a (yet unknown) degradation process,
thus ending its expression.
A refinement of this model, curiously never explored explicitly,
does not assume that nascent mRNA molecules enter ribosomes and start being
translated from their 5'-extremity. In contrast, it supposes that the 5'-triphosphate
end of the message folds back, and remains linked to RNA polymerase until
a specific signal, which can be located way downstream, tells it to detach
(and to terminate transcription). Antitermination has been thoroughly investigated
in the case of the antitermination protein N of bacteriophage lambda. This
process is readily compatible with a scanning process permitting the 5'-end
of the RNA to explore what happens in 3'-downstream sequences. The stringent
coupling of stable RNA synthesis to translation was also found to be linked
to the transcription elongation process. However, no clear-cut picture of
the control events of these processes are yet known. Since it is certainly
very difficult at this time to visualise in living cells the ongoing transcription
process, it will be interesting to look for 5'-3' correlations in the nucleotide
sequences of operons. This hypothesis thus places us back to the in silico
study of the genome text, demonstrating, once again, that, to understand
genomes it is necessary to go back and forth between the study of the physico-chemical
aspects of gene expression, and the formal study of the genome text. In summary,
one would expect two distinct fates for transcripts: either they would form
loops, with the 5' end scanning the 3' end until it encounters some termination
signal, or the 5' end would fold and form an RNA-protein complex, with specific
binding proteins, shifting away from the RNA polymerase transcribing complex.
This would be the case of the ribosomal RNA, that associate with ribosomal
proteins, but also of complexes such as the 5' terminal regulator of the transcription
control of tRNA synthetase genes in B. subtilis.
The study of the codon usage bias in bacterial genomes indicates
the presence of a strong selection pressure, which can only be understood
if one thinks that the corresponding genes are synthesized on ribosomes that
sit next to each other. The ribosome network organizes the cell's cytoplasm,
thereby providing the bulk of the mechanic forces needed to couple translation
transcription to the construction of the cell. A consequence of this interpretation
is that the position of the genes in the chromosome is not random. Ther must
exist a certain number of anchoring points which allow nascent transcripts
to couple transcription with traduction, and to make transcription of those
genes which are present in their immediate vicinity easy. It is quite possible
that several RNA polymerase molecules, yoked as draught animals by an appropriate
coupling factor, transcribe simultaneous several messenger RNAs corresponding
to products which have to be part of the same complex. As a consequence multimeric
proteins may be translated either from a single transcript (in an operon)
or from several transcripts synthesized next to each other. This means that
physically related functions have to be made from proteins which are synthesized
on ribosomes which sit in the vicinity of each other.
This raises an important question for genomics:
is it possible to find out, just knowing the genome text, whether a gene
product will form a protein complex? This is of course even more unlikely
than that an amino acid sequence could tell us exactly the fold of a
protein, without knowing pre-existing folds. Pancreatic RNase would fold
indeed, because selection isolated it with this behaviour (it is secreted
in bile salts), but this should never have been accepted, as it did,
as the paradigm of protein folding. Alignment with model proteins of
known structure will certainly help predicting a structure, because one
has to take into account the selective forces that have, during phylogeny,
led to the actual fold found in the models. This "threading"
approach is often used but it should be extended to the study of protein
complexes, by taking into consideration the intersubunit contacts in the
models. In
fact, the future of structural biology does not lie in the collection of
3D structures of all the proteins of a genome, as it is often proposed, but
in the identification of protein complexes, another example of the "neighborhood"
approach we advocate as a prelude to discovery.
Do we find a trace of this organisation in the chromosome?
It is illustrated, in the case of pathogenic bacteria, by the so-called pathogenicity
islands, where genes related to the function of virulence are grouped together.
And Agnieszka Sekowska and Eduardo Rocha have also recently demonstrated that
genes
involved in the metabolism of sulfur also make clusters, suggesting a
superorganisation of the corresponding gene products, probably due to the
fact that sulfur being a highly reactive atom, the gene products which deal
with it must be compartmentalized in the cell to protect it from the environment.
Another, more subtle, means of analysis is, once again, the study of the codon
usage bias in the genes. Ribosomes translating a mRNA with a highly biased
code will often require some tRNAs and rarely others. They will act as "attractors"
tending to fix the frequently used tRNAs in their vicinity. This local high
concentration will provide a positive selective advantage for all those mRNAs
which will have roughly the same codon usage bias, because they will be translated
rapidly. Therefore most mRNA molecules translated by these ribosomes will
tend to have the same bias: this is a further selective stabilisation of the
process. This phenomenon imposes that the same genes are often translated
at the same place in the cell, and therefore that their position in the genome
is fixed with respect to the general architecture of the cell. The cell is
thus viewed as a series of organised layers of ribosomes, as peels in onions,
where the concentration of tRNA varies progressively.
The main conclusion here is that all ribosomes are not equivalent
in the cell. But do we know exactly what is a ribosome? Their structure has
been determined in year 2000 — a truly remarkable technical feat —
and, as were the intuitions of Luigi Gorini in the ’70s, it has been
discovered that this was essentially an enzyme factory made of RNA, not of
proteins. Everything goes asif what we called ribosomes were the kernel of
a much more complex machine, with the translation initiation and elongation
factors, the tRNA synthetases, RNAs and other molecules making translation
work. This asks us to go back to the way used by biochimists to prepare ribosomes:
it is much similar to that used when one prepares cherries without kernels,
centrifugation. The kernel falls down during centrifugation, and the pulp
remains in suspension. What we name "ribosome" is the kernel, the
RNA core of much bigger objects, which are probably contacting each other
(which accounts for the fact that one observes en electron microscopy that
the ribosomes are regularly distributed along the messenger RNA, while they
are not in physical contact with each other), and of much more varied types.
During its translation, the messenger is like a thread going through a perl
necklace, where each perl has the same kernel, while its color varies: the
pulp of the ribosome would correspond to the local codon usage bias as well
as the presence of specific factors associated to the variety of compartments
in the cell, which varies according to the nature of genes. And, because helices
are universal forms (as we have seen), these necklaces of ribosomes are probably
arranged in helical structures which will make the core of the cell's organisation.
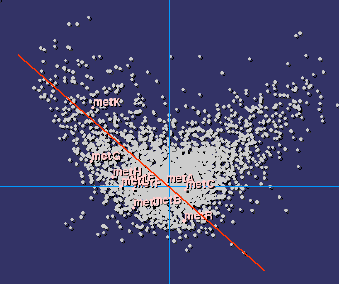 |
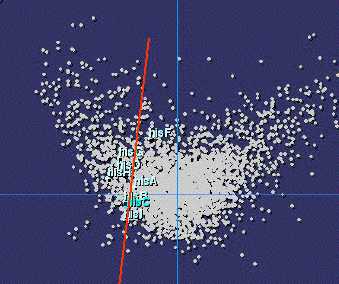 |
| Codon usage bias in methionine biosynthesis genes |
Codon usage bias in histidine biosynthesis genes (E. coli) |
It is therefore quite natural to study the distribution of
genes having a similar codon usage bias along the chromosome, and to relate
it with the metabolic or structural functions in the cell. We have found that
it is far from random: a linear correlation exists within each type of organised
metabolic pathway, showing that indeed there is a structural selective pressure
in the organisation of genes and gene products. The neighborhood approach
Indigo is meant to provide a first way to tackle this question, exploring
some of the most obvious neighborhoods.
A highly focused view, complementary to the one exposed
here is supported by the SIMEBAC
project of the Foundation
Fourmentin-Guilbert.
Training
and technology transfer
Integration of in vivo, in vitro and in
silico approaches require team work, and permanent conceptual technology
transfer from various disciplines and from various environments. Collaboration
between complementary views, such as the Greco-Latin, Anglo-American and
Chinese, is always beneficial. This is well understood by political structures
that we try to develop, for example through relevant collaborations (see
the European Focus for Biotechnology
in China). An important point is to make it properly understood that
technology transfer always require explicit acknowledgement of the sources
of the technology, be they conceptual or technical. Since the end of 2001,
a working seminar has been initiated at the Department of Mathematics of
the University of Hong Kong (Pr Ngaiming Mok) where students and scientists
from the Hong Kong region mixed up around Danchin and Mok on a reflection
about Conceptual Biology. This seminar has resumed at the Institut Pasteur
in Paris, on a less regular basis. An account of each meeting is sent to
the participants as well as to all those belonging to the Stanislas Noria
network (Causeries du jeudi).
|
|
|
| (2)...... to create a culture of
basic research allowing scientists to monitor emerging diseases surveillance,
prevention and cure.... |
 |
(1) .... to create
knowledge- and education-related information resources based on laboratory
experiments and "in silico" analysis ..... |
|
(3) ..... to offer new areas
for technology development used in large-scale industrial applications,
fostering future research....... |
|