Project list
Neuronal activity in the Cerebellar Nuclei in freely moving mice (on going)
Sleep staging in calves (on going)
DISK: imputing missing data from 2D and 3D keypoint tracking (on going)

Understanding immune-cancer relationships from multiplexed images
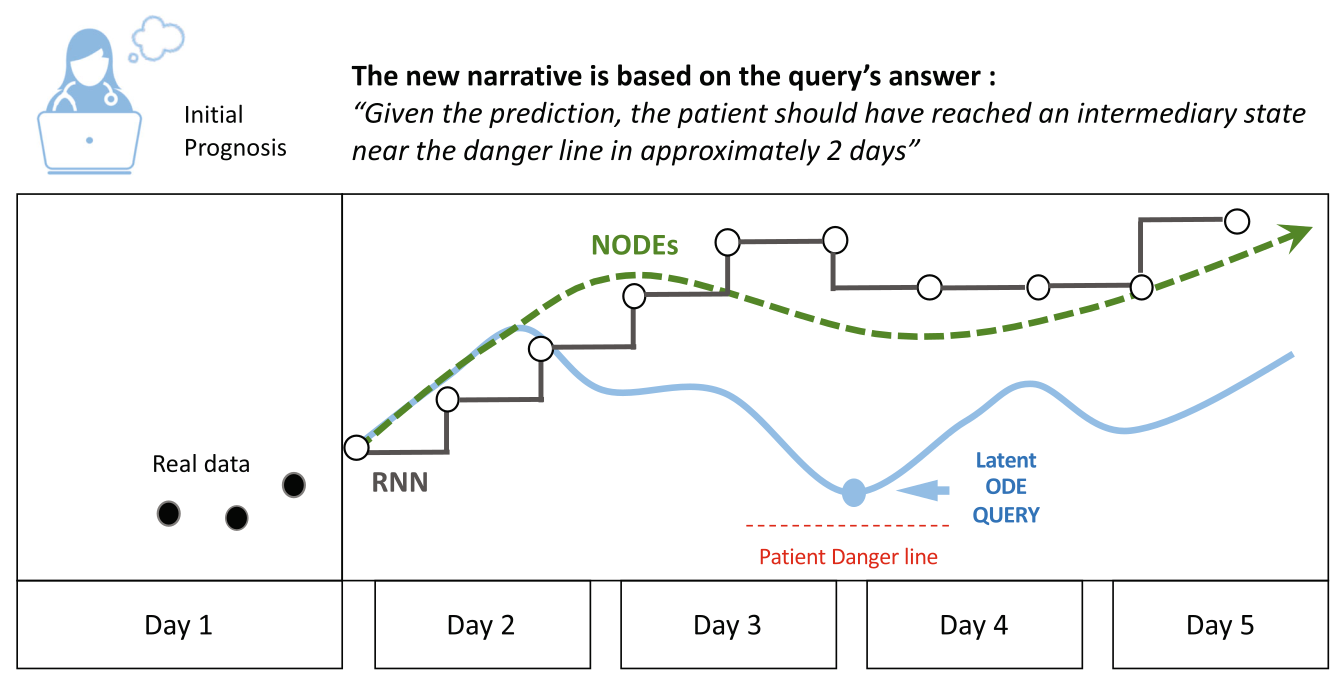
Can visualizing forecast foster human trust for machine learning-based medical prognosis?
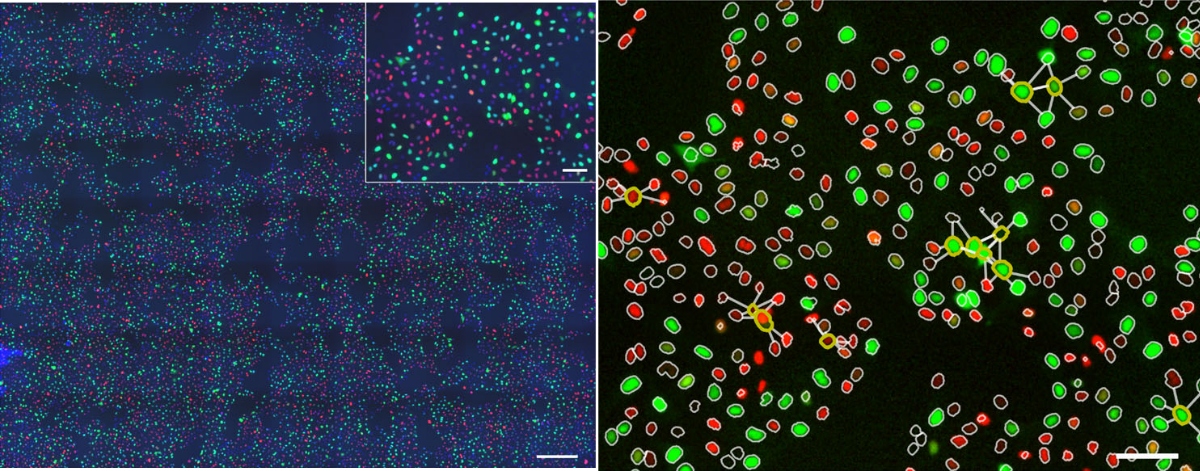
pySpacell: a Python package for single-cell spatial image analysis
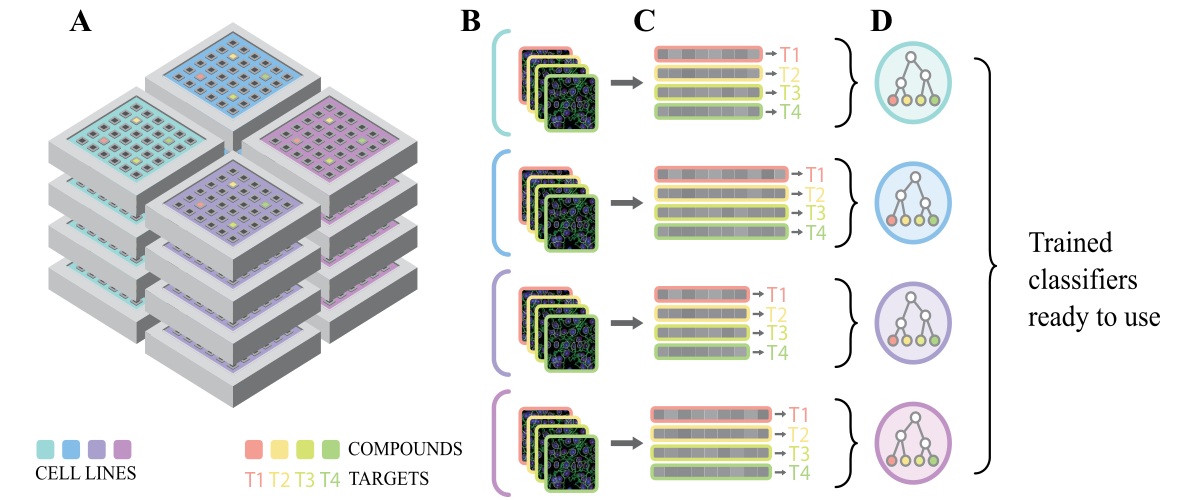
Using multiple cell lines for drug target prediction in high-content screening

Unsupervised characterization of cellular heterogeneity in cell culture

Discovering genes responsible of oriented cell division with high-throughput microscopy videos
Representations of animal movements in long sequences (on going)
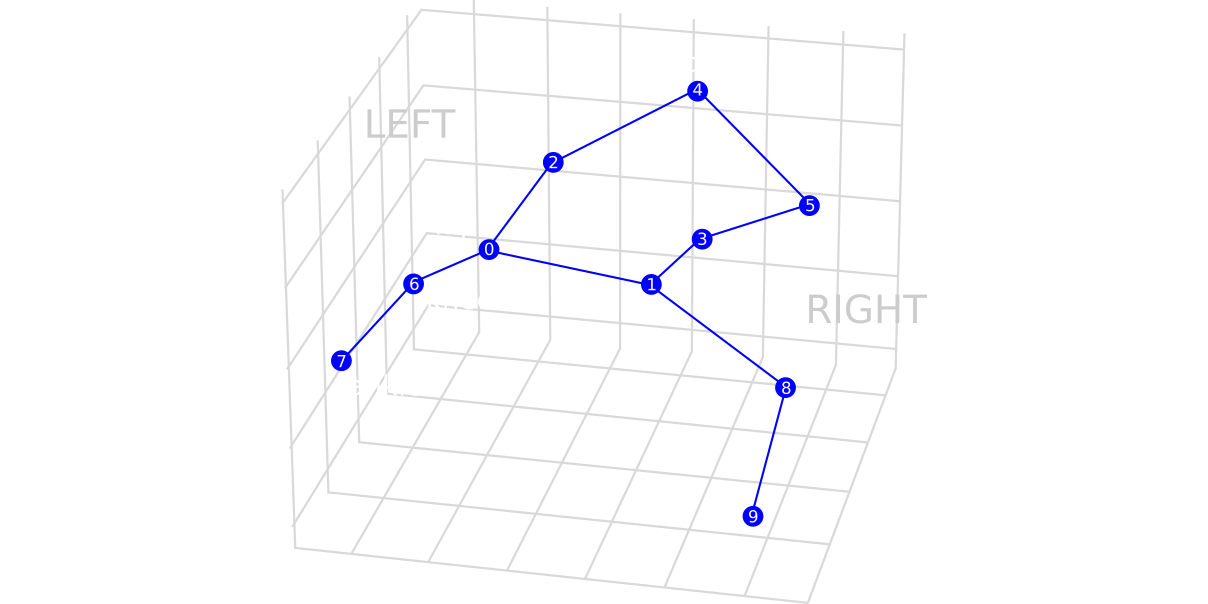
Animal behavior provides a non-invasive readout of underlying neural activity: even subtle changes in movement can reflect neurological alterations induced by internal or external factors, such as genetic variation or pharmacological treatments. We aim at identifying these changes by modeling the behavioral dynamics, i.e. by classifying long sequences of skeleton-based pose data.
Rather than relying on hand-crafted features, we are developing a classifier that is both accurate and interpretable, capable of objectively identifying discriminative behavior patterns that differentiate experimental conditions.
We first use a masked autoencoder (MAE) based self-supervised learning method — commonly used in computer vision — as a pretraining backbone to extract frame-level representations. We then use an aggregation technique to obtain sequence-level features for classification. This way, we can handle long sequences, providing a richer biological insight. We think this approach will lead to a better understanding of how normal behaviors (e.g. walking, balance, orientation in space, and very subtle motion changes) are impaired under specific pharmacological treatments.
Keywords: behavior, representation learning, transformers, multiple instance learning (MIL)
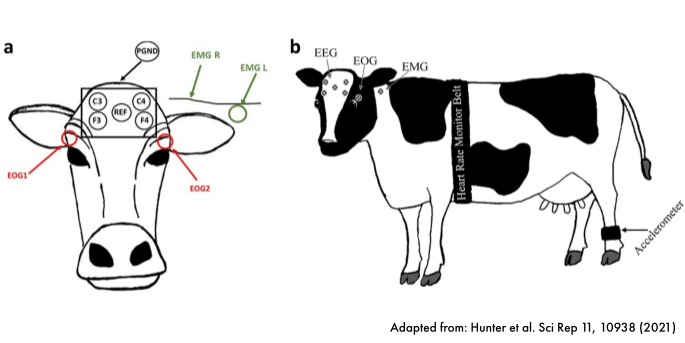
Sleep staging in calves (on going)
In collaboration with Prof. Dr. Jenny Stracke (University of Bonn), we are developing new automatic pipelines to detect sleep segments and sleep stages (wake, light sleep, deep sleep, REM sleep) using different physiological signals (muscle activity, brain activity, eye movements, heart rate). Sleep staging tools based state-of-the-art polysomnography or wearable data sources are for humans available. However tools achieving same results in other species are lacking. Reliable tools for sleep staging for cattle will open doors to more behavior quantification including intra-/inter-individual variability.
Keywords: behavior, sleep staging, EEG, farm animal
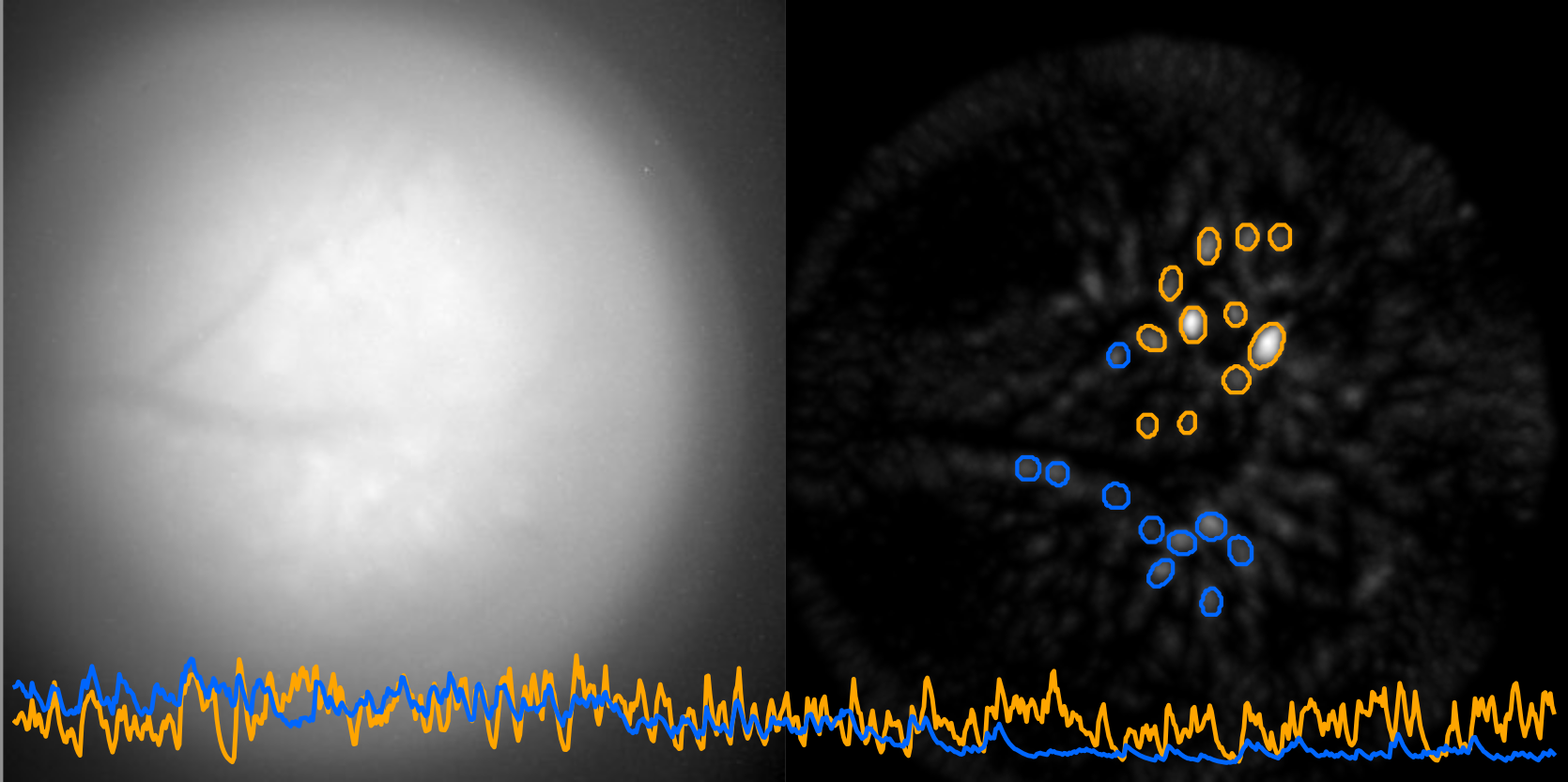
Neuronal activity in the Cerebellar Nuclei in freely moving mice (on going)
Imaging deep brain structure activity in a freely-behaving mouse remains an experimental challenge. Here, our collaborator Dr. Bogna Ignatowska-Jankowska (Okinawa Institute of Science and Technology) and her colleagues, used a miniature endoscope affixed on the mouse head to visualize the neuronal activity through calcium imaging in the cerebellar nuclei. Together, we aim at comparing the activity of the same neurons across distinct behavioral tasks to shed light on the role of cerebellar nuclei in the control of behavior.
Keywords: behavior, in-motion neural recordings, cerebellar nuclei, deep brain structures
DISK: imputing missing data from 2D and 3D keypoint tracking
Both video recordings coupled with automated posture estimation and motion capture systems are producing high quality data of an individual’s motion. Yet these methods are not perfect and contain missing data. We developed Deep Imputation for Skeleton data (DISK), a deep learning algorithm to learn dependencies between keypoints and their dynamics to impute missing tracking data. We demonstrated the usability and performance of our imputation method on six different animal skeletons including two multi-animal set-ups.
Keywords: behavior, representation learning, transformers, missing data, data imputation
Other resources:
Coupling DISK with pose estimation algorithms (on going)
Markerless pose estimation methods such as DeepLabCut and SLEAP learn in a supervised fashion to predict the locations and the confidence score of user-defined keypoints from videos. To achieve high tracking accuracy, not only good lighting and viewpoint conditions are required, but also extensive manual annotations.
In this project, we aim to show that DISK can reduce the requirement for labeling while achieving equivalent tracking accuracy.
DISK by learning the movement dynamics could complement pose estimation algorithms that are only working on static individual images. DISK will impute sequences that are deemed of poor tracking quality. To this aim, we are exploring methods to filter pose estimation outputs, as current approaches do not always correlate with actual prediction errors. Our goal is to create an automated pipeline that will allow researchers to streamline animal tracking through combining pose estimation tools and imputation of missing data with DISK.
Keywords: behavior, pose estimation, markerless tracking, transformers, neural network calibration, data imputation
Towards a foundational behavior model (on going)
Building on the success of the self-supervised learning-based DISK framework, we explore the transferability of core behavioral patterns shared within and across species. We are testing different neural network architectures that offer scalability — following trends of deep learning foundation models — and transferability to unseeen datasets. In a parameter-efficient manner, we seek to enhance DISK's behavioral profiling and data imputation capabilities, ultimately driving toward a universally generalizable model for animal behavior analysis.
Keywords: behavior, foundational model, transformers, missing data, data imputation, generalizability across species
Understanding immune-cancer relationships from multiplexed images
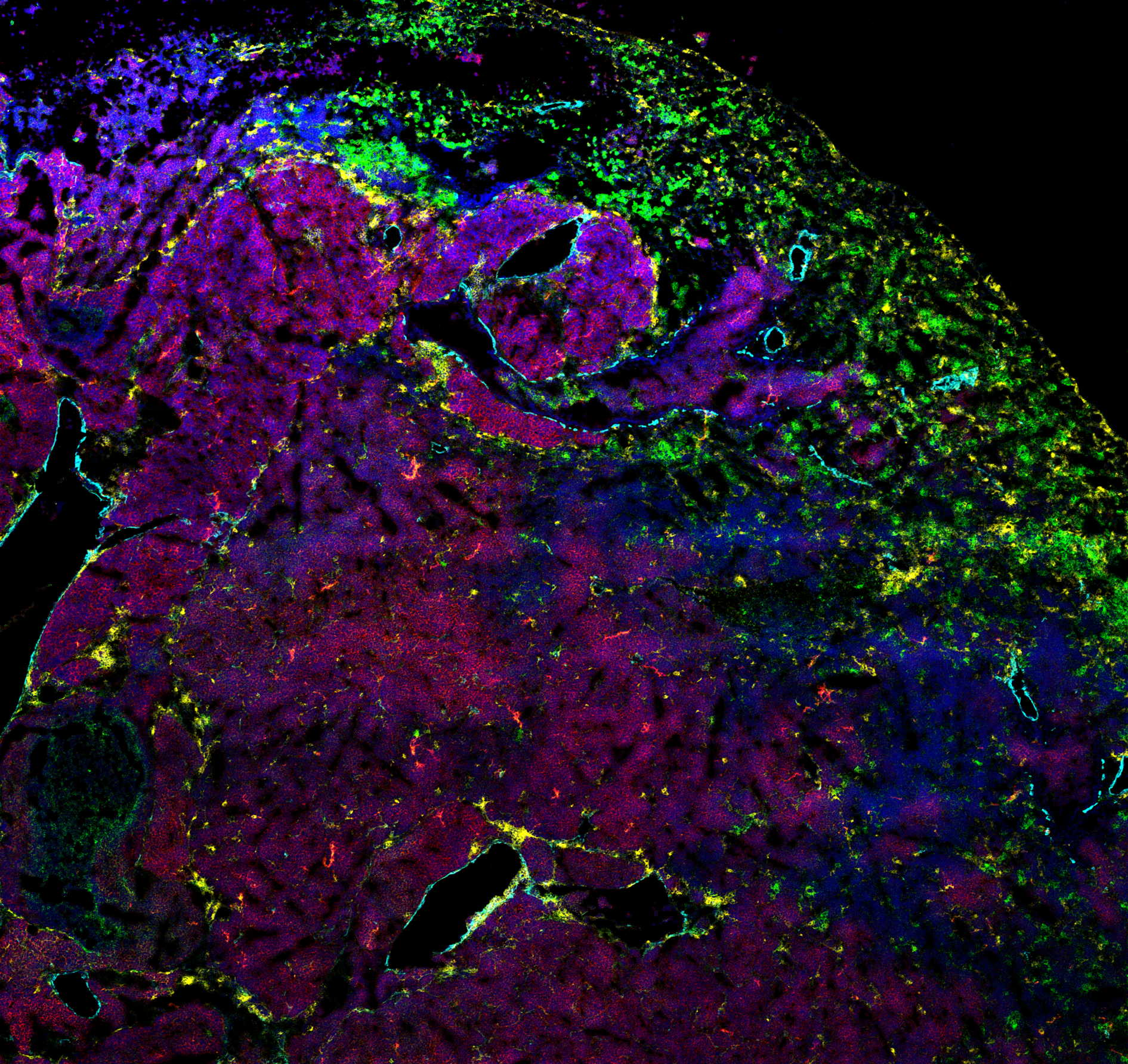
Traditional fluorescence microscopy is limited by spectral overlap: multiple fluorophores can present an overlap in their exciting wavelength ranges, making it impossible to separate signals. The spectral overlap limits to a handful the number of fluorophores that can be imaged at one point. A few techniques have pushed this limit. Mass-Spectrometry Imaging is using metal beads attached to antibodies to "image" up to 50 proteins at a time. A powerful laser burns a small piece of the sample (1 squared micrometer) and the volatile metal beads are recognized with a mass-spectrometer. Mass-spectrometry imaging is a very promising imaging technique to analyze the spatial organization of a tissue while having access to an unprecedented number of proteins.
We use mass-spectrometry images to analyze the tumor micro-environment of two types of cancer: small cell-line lung cancer (SCLC), and Chronic Lymphocytic Leukemia (CLL). We investigate the infiltration of immune cells in the tumor, the changes of protein expressions in different compartments, and the effect of advanced cancer treatments.
From the analysis point of view, the detection and segmentation of cells is difficult due to the low resolution of mass-spectrometry images (1 squared micrometer) compared to traditional microscopy. We develop crafted methods to detect all relevant celltypes, with special care for rare cell types. we further compare the protein expressions and tissue architectures.
Keywords: MIBI-TOF, imaging mass spectrometry, multiplex imaging, segmentation, tumor micro-environment
Articles:
Can visualizing forecast foster human trust for machine learning-based medical prognosis?
Machine learning (ML) has recently been demonstrated to rival expert-level human accuracy in prediction and detection tasks in a variety of domains, including medicine. Despite these impressive findings, a key barrier to the full realization of ML’s potential in medical prognoses is technology acceptance. Recent efforts to produce explainable AI (XAI) have made progress in improving the interpretability of some ML models, but these efforts suffer from limitations intrinsic to their design: they work best at identifying why a system fails, but do poorly at explaining when and why a model’s prediction is correct. We posit that the acceptability of ML predictions in expert domains is limited by two key factors: the machine’s horizon of prediction that extends beyond human capability, and the inability for machine predictions to incorporate human intuition into their models.
We propose the use of a novel ML architecture, Neural Ordinary Differential Equations (NODEs) to enhance human understanding and encourage acceptability. Our approach prioritizes human cognitive intuition at the center of the algorithm design, and offers a distribution of predictions rather than single outputs. We explain how this approach may significantly improve human-machine collaboration in prediction tasks in expert domains such as medical prognoses. We propose a model and demonstrate, by expanding a concrete example from the literature, how our model advances the vision of future hybrid human-AI systems.
Keywords: Neural Ordinary Differential Equations (NODEs), XAI, medical prognosis
pySpacell: a Python package for single-cell spatial image analysis
Technologies such as microscopy, sequential hybridization, and mass spectrometry enable quantitative single-cell phenotypic and molecular measurements in situ. Deciphering spatial phenotypic and molecular effects on the single-cell level is one of the grand challenges and a key to understanding the effects of cell–cell interactions and microenvironment. However, spatial information is usually overlooked by downstream data analyses, which usually consider single-cell read-out values as independent measurements for further averaging or clustering, thus disregarding spatial locations. With this work, we attempt to fill this gap. We developed a toolbox that allows one to test for the presence of a spatial effect in microscopy images of adherent cells and estimate the spatial scale of this effect.
The proposed Python module can be used for any light microscopy images of cells as well as other types of single-cell data such as in situ transcriptomics or metabolomics. This toolbox allows to test for the presence of a spatial effect in microscopy images of adherent cells and estimate the spatial scale of this effect. It can be used for any light microscopy images of cells as well as other types of single-cell data such as in situ transcriptomics or metabolomics. The input format of our package matches standard output formats from image analysis tools such as CellProfiler, Fiji, or Icy and thus makes our toolbox easy and straightforward to use, yet offering a powerful statistical approach for a wide range of applications. The available spatial tests are available for both categorical and continuous cell features.
Keywords: spatial analysis, single-cell, statistical test, python
Using multiple cell lines for drug target prediction in high-content screening
In phenotypic cell-based assays, drugs are directly tested onto cells to assess their effect. Using cells allow scanning a wide spectrum of possible drug targets at once. Cell-based assays proved to be efficient at discovering first-in-class therapeutic drugs. However, posterior identification of a drug's mechanism of action (MOA) has remained difficult and highly refractory to automated analyses. Methods increasing the number of fluorescent dyes to reveal relevant cellular components were suggested for MOA prediction. We demonstrated that adding fluorescent dyes to a single assay has limited impact on MOA prediction accuracy, as monitoring only the nuclei stain could reach compelling levels of accuracy. This observation suggested that multiplexed measurements are correlated and nuclei stain could possibly reflect the general state of the cell. We then hypothesized that combining unrelated and possibly simple cell-based assays could be used to predict a drug target. We trained an ensemble classifier to predict drug targets and prioritize a possibly large list of unknown compound hits at once. Moreover, we show that such a combination of past screen data is usally found in screening facilities and can be re-used without additional experimental costs.
Keywords: High-Content Screening (HCS), random forests, ensemble classifier
Unsupervised characterization of cellular heterogeneity in cell culture
Robotics and automated fluorescence microscopes have promoted high-content cell-based screenings: fluorescent probes targeting DNA or other cell components are used to image hundreds of thousands of cells under many different conditions. Cell-based assays have proven to be efficient at discovering first-in-class therapeutic drugs, i.e. drugs acting on a new target. They allow to detect promising molecules and to associate functional annotations to them, like their molecular target or mechanism of action (MOA).
Even clonal cells respond differently under the same treatment. I studied this heterogeneity and its impact on drug profiling. Clustering approaches can be used to uncover cell subpopulations. To evaluate the additional information brought by the subpopulation approach, I compared the performances of clustering on a MOA prediction. I used an open-source dataset where 38 drugs were tested on a breast cancer cell line and imaged with three fluorophores targeting DNA, Actin and Tubulin (BBBBC021 from the Broad Institute). I additionally tested the reproducibility of the clustering algorithms. For both performance and reproducibility, the PhenoGraph (or Louvain) algorithm was the best choice.
Additionally the modelization was enriched with the spatial organization of cell subpopulations. I found that neighboring cells influence each others, and display a similar phenotype more frequently than expected at random. These results assessed across a hundred of treatments, show that even genetically identical cells are not all alike and independent, but create spatial heterogeneity via cell lineage and interaction. Using spatial information as well as phenotypic heterogeneity with graph kernel methods improves the MOA classification under some conditions.
Keywords: High-Content Screening (HCS), mechanism of action (MOA), clustering, graph kernels
Discovering genes responsible of oriented cell division with high-throughput microscopy videos
Coated micropatterns on slides allow individual growing cell clusters at large scale. Combined with automated microscopy, tens of thousands of videos of these growing cell clusters could be recorded. These videos can help identifying factors involved in cell growth, cell division or tissue formation by testing multiples perturbations (genetic or drug). The focus of the video analysis was the division angle from 2 to 3 cells and which perturbations caused a change in the division angle distribution.
However, cells growing on a micropattern tend to be tightly packed and to overlap with each other. The image analysis of those large dynamic datasets with no possible human intervention is particularly challenging and has proven impossible using out-of-the-box automated cell detection methods.
We proposed a fully automated image analysis approach to estimate the number, the location and the shape of each cell nucleus, in clusters at high throughput. The method is based on a robust fit of Gaussian mixture models with 2 and 3 components on each frame followed by an analysis over time of the fitting residual and two other relevant features. We used the time resolved analysis to identify with high precision the very first frame containing three cells. We demonstrate the accuracy of our method by validating it against manual annotation on about 4000 videos of cell clusters.
Keywords: Gaussian mixture, high-throughput, time-lapse microscopy, cell detection