
Who has never been lost during simulation development or data analyses into the myriad of different parameters, result folders and figures produced by wandering around the research path?
Maybe your supervisor asked you to see if results change with this new set of parameters? Or maybe you want to try out what happens if you remove migration between two populations?
You have your nice Snakemake pipeline that automates the production of results, you can change parameters easily. However, once you do, even if you keep track of changes with version control, your previous results/figures will be overwritten unless you spend a good amount of time designing your pipeline so that all output files or folders have a unique name reflecting the parameter set. This is painfully tedious and you end up with something like results/sim_t12_d8_s0.001_N10000_seed2837283.svg.
So I set out to do a little experiment and find a way to keep parameters info and outputs neatly organized for exploring around.
I don’t want to commit the results to the git directory, this would work in this small example but not be very practical with often very large analyses and simulations projects.
The following example requires some familiarity with git.
A simple example simulation
Let’s first build a simple simulation and output figure using Snakemake and msprime (a coalescent simulator for ancestral histories and DNA sequence data). I’m biased by my field here, which is population genetics.
This little experiment requires python, Snakemake and msprime (which is a python module). All installable through conda or pip for example.
mamba create -n sim_pipeline python bioconda::snakemake conda-forge::msprime
# or
pip install snakemake msprimeLet’s create a folder for our experiment and initialize a git repository.
mkdir sim_pipeline
cd sim_pipeline
git init
git branch -m master main # yeah the default 'master' is pretty bad...Now we build a simple Snakefile that will produce an svg figure of a tree sequence produced by msprime:
Snakefile [main]
rule all:
input:
f"results/sim.svg",
rule simulation:
output:
svg = f"results/sim.svg",
params:
seed = 1246682,
samples = 5,
rec = 1e-8,
N = 5_000,
run:
import msprime
ts = msprime.sim_ancestry(
samples=params.samples,
recombination_rate=params.r_rec,
sequence_length=10_000,
population_size=params.N,
random_seed=params.seed,
)
ts.draw_svg(output.svg)Running the pipeline with
snakemake --cores 1will create the file results/sim.svg 
Let’s add our pipeline file and commit it.
git add Snakefile
git commit -m 'create Snakefile'Creating outputs based on the git branch name
In this section, we will add some code that will name the output image according to the git branch we are in (but this could also be according to commit hash for example).
Let’s first create a python function in the Snakefile that returns the branch name (here prefixed with an underscore by default) and add that to our output name.
Snakefile [main]
import subprocess
import warnings
def get_git_branch(prefix='_'):
try:
return prefix + subprocess.check_output(['git', 'branch', '--show-current']).strip().decode('ascii')
except:
warnings.warn('probably not a git repository')
return ''
rule all:
input:
f"results/sim{get_git_branch()}.svg",
rule simulation:
output:
svg = f"results/sim{get_git_branch()}.svg",
params:
seed = 1246682,
samples = 5,
rec = 1e-8,
N = 5_000,
run:
import msprime
ts = msprime.sim_ancestry(
samples=params.samples,
recombination_rate=params.r_rec,
sequence_length=10_000,
population_size=params.N,
random_seed=params.seed,
)
ts.draw_svg(output.svg)This time the command
snakemake --cores 1will create the file results/sim_main.svg (same image as above but with different name)
Let’s finally commit those changes, we wouldn’t want to lose them.
git commit -am 'name output according to branch'Changing parameters
We are ready to explore our parameters (they are in the simulation rule, params attribute). Let’s change the seed (seed), sample number (samples), recombination rate (rec) and population size (N).
We create another branch called params1 and switch to it
git checkout -b params1Now we can change the parameters Snakefile and commit it.
Snakefile [params1]
...
rule simulation:
output:
svg = f"results/sim{get_git_branch()}.svg",
params:
seed = 8468734,
samples = 6,
r_rec = 2e-8,
N = 3_000,
...git commit -am 'params1 parameter set'Running the pipeline as above we obtain a new output called results/sim_params1.svg 
Great! We now have an output, with a corresponding history of the changed parameters.
With git, it is also pretty easy to find the differences between two branches without having to change between them:
git checkout main
git diff main params1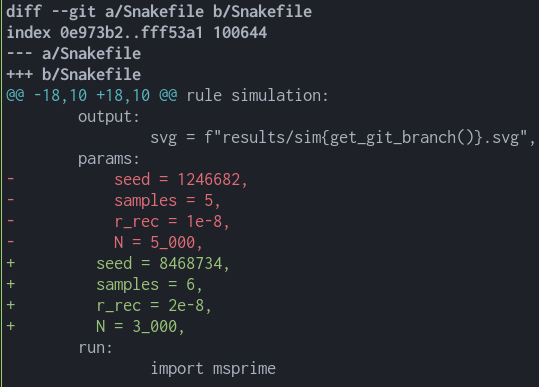
Using a main config file in larger projects
Managing a lot of parameters can quickly become complex in larger projects. Snakemake offers the possibility to have a general config file in YAML or JSON.
We can create a new file named config.yaml
config.yaml
simulation:
params:
seed: 1246682
samples: 5
r_rec: 1e-8
N: 5_000And modify the Snakefile to make use of this config
Snakefile [main]
import subprocess
import warnings
1configfile: "config.yaml"
...
rule simulation:
output:
svg = f"results/sim{get_git_branch()}.svg",
params:
2 **config['simulation']['params']
run:
import msprime
ts = msprime.sim_ancestry(
samples=params.samples,
recombination_rate=params.r_rec,
sequence_length=10_000,
population_size=params.N,
random_seed=params.seed,
)
ts.draw_svg(output.svg)- 1
- import the config file, the yaml is parsed into a python dictionary
- 2
- use python’s dictionary unpacking to have all the params available for use in this rule
In the end, I don’t know how useful it is going to be to me but at least I now have this option in my toolbox.
Citation
@online{simon2023,
author = {Simon, Alexis},
title = {Keeping Track of Your Simulation Parameters with {Snakemake}
and Git},
date = {2023-06-30},
url = {https://www.normalesup.org/~asimon/posts/2023-06-30-snakemake-git-simulations/},
langid = {en}
}