The automat2 software user guide
Required data formats
automat2 inputs biological sequence banks in the widely used FASTA format. in this format, the databank
file is in ASCII (editable) text.
Each sequence starts with a single line header starting with a ``>'' sign in the leftmost column,
followed by a unique sequence identifier, a separating space and other data.
From the automat2 viewpoint, everything right to the ``>'' character and the sequence identifier is
sequence comment (in practice it contains sequence length and other ancillary info).
The following lines (up to the next ``>'' starting line or the end of file) contain the sequence
possibly interlaced with blank characters (space, tabs or newlines).
Here is an extract of a FASTA format file:
>gb|AJ532906|AAL532906 Alces alces kappa-casein gene, exon 4, isolate f3.
AAAATAGTCAAATATATCCCAATTCAGTATGCGCTGAGTAGGTATCCTAGTTATGGACTC
AGTTACTACCAACACAGACCAGTTGCACTAATTAATAATCAATTTCTGCCATACCCATAT
TATGCAAAGCCAGGTGCAGTTAGGTCACCTGCCCAAATTCTCCAATGGCAAGTCTTGCCA
AATACTGTGCCTGCCAAGTCCTGCCAAGCCCAACCAACTACCATGGCACGTCACCCACGC
CCACGTTTATCATTTATGGCCATTCCGCCAAAGAAAAATCAGGATAAAACAGACATCCCT
ACCATCAATACCATTGCTACTGTTGAGTCTACAATTACACCCACCACCGAAGCAATAGAG
GACAATGTAGCTACTCTAGAAGCTTCCTCAGAAGTTATTGAGAGTGCACCTGAGACCAAC
ACAG
>gb|M98484|AALMTCYTOB Alces alces americana cytochrome b gene,
partial cds; mitochondrial gene for mitochondrial product.
ACTTCGGTTCTCTATTAGGAGTTTGCTTAATCTTACAAATCCTTACAGGACTATTCCTAG
CAATACATTATACACCCGACACAATAACAGCATTCTCCTCTGTCACCCACATCTGCCGAG
ATGTAAATTACGGCTGAATCATTCGATATATGCATGCAAACGGAGCCTCAATATTCTTCA
...
Though virtually every biological databank are already available in FASTA format, the two gp2fasta and
swissprot_to_fasta programs make conversion from two other popular formats.
Automat2 makes also use of two other proprietary (but trivial) data formats: The first one is an
alphabet file, which is an editable file contening on each line all the sequence symbols (database
letters, i.e. residues or nucleotides) that are considered as identical by the match search algorithm
(the ``automaton'' to the proper sense). An example could be for proteins:
VILM
RQEKZ
HSDB
FYW
ACT
GN
P
Note that we did not have a line for unknown residuals (generally denoted X in protein databanks and
N in genetic databanks). Indeed, the unreferenced symbols break any sequence match, while adding the
X (resp.N) symbol in the alphabet file would make unknown sequence symbol match the filtered out query
sequence symbols (cf the ``query sequence filters'' section below), which is not a very sound idea!
The second format is for the symbol distance matrix used in the match scoring (such as Blosum or
Dayoff). The first line of the file contains the symbols in the matrix row order, and the following
lines contain each one single row of the matrix (with entries separated by blank characters).
Since this matrix is generally symetrical (otherwise it is NOT a distance, though automat2
may function as well with nonsymetrical matrices) we may only provide the lower triangular terms.
Here is the content of a matrix for proteins (Dayoff's):
CSTPAGNDEQHRKMILVFYWX
120
0 20
-20 10 30
-30 10 0 60
-20 10 10 10 20
-30 10 0 -10 10 50
-40 10 0 -10 0 0 20
-50 0 0 -10 0 10 20 40
-50 0 0 -10 0 0 10 30 40
-50 -10 -10 0 0 -10 10 20 20 40
-30 -10 -10 0 -10 -20 20 10 10 30 60
-40 0 -10 0 -20 -30 0 -10 -10 10 20 60
-50 0 0 -10 -10 -20 10 0 0 10 0 30 50
-50 -20 -10 -20 -10 -30 -20 -30 -20 -10 -20 0 0 60
-20 -10 0 -20 -10 -30 -20 -20 -20 -20 -20 -20 -20 20 50
-60 -30 -20 -30 -20 -40 -30 -40 -30 -20 -20 -30 -30 40 20 60
-20 -10 0 -10 0 -10 -20 -20 -20 -20 -20 -20 -20 20 40 20 40
-40 -30 -30 -50 -40 -50 -40 -60 -50 -50 -20 -40 -50 0 10 20 -10 90
0 -30 -30 -50 -30 -50 -20 -40 -40 -40 0 -40 -40 -20 -10 -10 -20 70 100
-80 -20 -50 -60 -60 -70 -40 -70 -70 -50 -30 20 -30 -40 -50 -20 -60 0 0 170
-80 -80 -80 -80 -80 -80 -80 -80 -80 -80 -80 -80 -80 -80 -80 -80 -80 -80 -80 -80 -80
The presence of a line for symbol X should not be confusing, since sequence filters (see the
``query sequence filter'' section below) change the discarded residuals to X in the protein mode and to
N in the nucleic acid mode. The corresponding matrix line defines how to score such filtered out
subsequences in the match listing.
The search parameters
Automat searches the sequences in three phases (although for computational efficiency all three are made
simultaneously in an interleaved way):
First the search automaton detects streaches, above a given ``triggering'' length, of consecutives
symbols, identical for the searching alphabet between the query and the databank sequence.
When all these ``primer'' streaches are found, the algorithm gathers successive primers (under a
distance threshold) and counts the number of identical symbols (optionally taking into account identical
symbols in between and around the primers). If the total number of identical symbols is above a given
threshold, the match is scored.
The scoring by the user-provided comparison matrix finds the sub-interval of (a neighborhood of) the
match that gives the best summed difference matrix entries between the query sequence symbol row and
the databank sequence symbol column.
For a given sequence, only the few best matches are stored (their number is user defined) provided
the score is high enough (above an user-defined threshold).
Eventually, the first few best sequences are reported, sorted by decreasing score. In each sequence
report, the matches are also sorted by decreasing order (with the exception that successive matches
with the same offset are reported together). Neighboring matches are merged for display constraints.
Since only a given number of sequences are reported, (and respectively a given number of matches per
sequence), there is the option of discarding all matches of which score if lower than the score of
unreported sequences (resp. sequences of which score -i.e. best match score- is lower than the score
of unreported matches of reported sequences).
Here is an annotated screenshot of automat2X11 showing the search parameters effects :
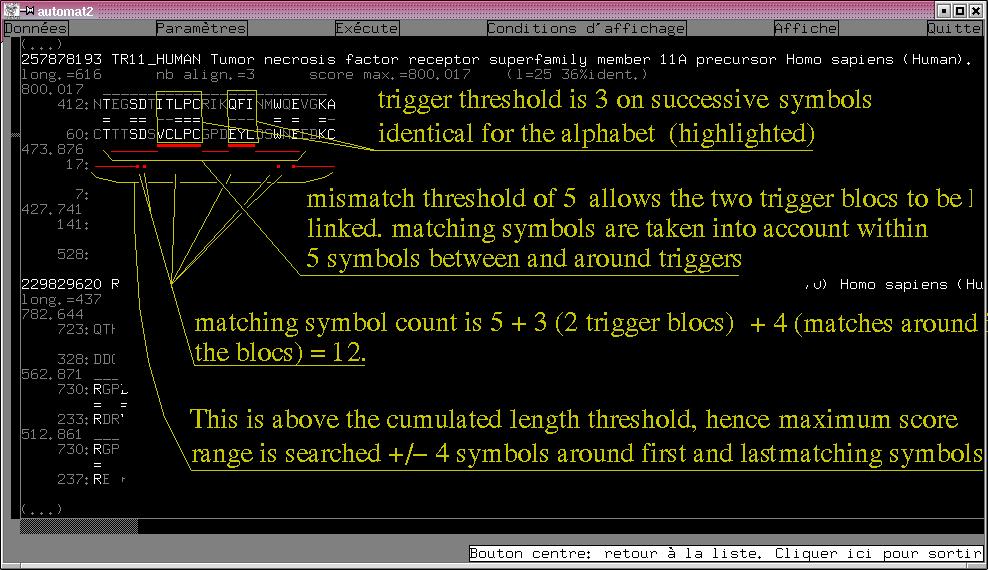
parameters are : Protein mode, Alphabet given above, Dayoff matrix given above, Matrix dithering on,
Trigger length 3, Cumulated length threshold 7, Mismatch length 5, Take matching symbols into account
around triggers on.
Sequence score is unlimited, Max number of reported sequences=1000, Matches below unreported sequences
score are not discarted, Max number of matches per sequence=3, Sequences below unreporetd
matches are not discarded.
In case the matching symbols are not taken into account between and around the trigger blocs,
the count would have been 5+3 = 7 above the cumulated length threshold, but the maximum scoring range
would have been searched on a shorter 19-residue long interval (+/-4 symbols around the triggers).
extractor, a FASTA sequence extraction tool
Since query sequence(s) are also read from a fasta file, it may be usefull to extract a sub-databank
from the sequence comment contents in order to make query selection easier. The extractor program allows
to select from a FASTA database a subset based on the presence/absence of given words in teh comment.
The program is invocated with two arguments: First a databank to be read from, and second a databank
subset file to be created. Then a dialog (in French, but I will give you the meanings) begins, in which
the user is prompted for a word (succession of characters that will be searched for irrespective of the
case) on a line (an empty line means ``everything'') separated by connexion lines. Connexion lines are
any of the following French expression:
| Expression |
What it means |
| et |
AND |
| et pas |
AND NOT |
| ou |
OR |
| mais pas |
BUT NOT |
| |
(empty line finishes the input) |
The program asks then whever it should list the comments of the extracted sequences. Answer by either
``oui'' (yes) or ``non'' (no)
Here follows an example of extraction (underlined text have been input by the user):
#extractor swissprot-40.fasta apoptosis.fasta
Entrer les chaînes à rechercher dans les en-têtes, séparées par des lignes "et", "et pas", "ou" ou "mais pas"
La ligne de séparation est vide aprés la dernière chaîne à rechercher.
La comparaison ne tient pas compte des majuscules minuscules.
Une ligne vide correspond à "tout".
apoptosis
et
human
(this is an empty line)
0 APOPTOSIS
2 HUMAN
listage des séquences extraites ?
oui
On y va...
15366331 APR2_HUMAN Apoptosis related protein APR-2. Homo sapiens (Human).
18310967 ASC_HUMAN Apoptosis-associated speck-like protein containing a CARD (hASC) Homo sapiens (Human).
24419231 BAK2_HUMAN Bcl-2 homologous antagonist/killer 2 (Apoptosis regulator BAK-2). Homo sapiens (Human).
24422555 BAK_HUMAN Bcl-2 homologous antagonist/killer (Apoptosis regulator BAK). Homo sapiens (Human).
...
211122200 REQU_HUMAN Zinc-finger protein ubi-d4 (Requiem) (Apoptosis response zinc finger Homo sapiens (Human).
257889198 TR12_HUMAN WSL-1 protein precursor (Apoptosis-mediating receptor DR3) Homo sapiens (Human).
22 séquences extraites.
Last line reports the number of extracted sequences.
query sequence filters
It may be sometimes usefull to discard unrelevant parts of the query sequence (for example, parts
of which information content is low).
Automat is provided with some query sequence filters, but you may create your own.
The filters transforms the sequence read as input by adding 128 to the symbol code of the discarded part
and outputs the resulting code.
As generally sequences contains only uppercase letters (ACDEFGHIKLMNPQRSTVWY for protein, ACGTU for
nucleic acids) adding 128 raises the higher bit of the character resulting into an invisible one (or an
accentuated character if the terminal is set up for non-ASCII charset). The automat2X11 GUI displays
such filtered out symbols as dimmed, the user can manually change the status of an interval in the query
sequence by pressing the mouse button at the begining of the interval and release the button at the end
of the interval.
Available filters are:
nullfilter
this filter makes nothing! This seems silly, but it is very usefull for manually deselect areas in
your query sequence(s) with the automat2X11 GUI (specifying this filter pops up a sequence display on
which you can deselect areas with the mouse buttons).
polyAfilter n
This filter discards subsequences of n or more identical symbols.
HFRfilter n1 n2 ... nk
This filter discards high frequency repeats of periods 1 up to k of length
n1 n2 ... nk respectively (thus HFRfilter n is
equivalent of polyAfilter n). The constraint is that period is meaningfull that mean that
the pattern is repeated at least twice (i.e. np ≥ 2p).
lowHfilter bank.inf n b [alphabet]
This filter discards subsequences of low informational content (entropy). Its arguments are:
- first the info file generated by automat2 the first time it scans a databank file (this info filename
has a .inf extension),
- second the length n of analysis,
- third a threshold on the number of bits equivalent to the length of analysis,
- and optionally a fourth argument specifying an alphabet used in the sequence analysis. This alphabet
file contains lines equivalent symbols, possibly followed by a space character and a list of composing
symbols. For example (for DNA/RNA sequences):
Aa
Cc
Gg
TtUu
Nn ACGTacgtUu
Note 1: alphabet files of automat2 may be used as argument, but non conversely, in the example above,
the last line format is not compatible with automat2 (it means that letter N in the databank stands for
an unknown base).
Note 2: it is important to know that this program does not take into account the order of the
symbols but only their local abundances. Thus, this filter is generally uninteresting for nucleic acids
(despite of my alphabet example, but an example with 20 symbols wouldn't be as illustrative).
Filters effects can be cumulated by piping then one to the other. For example:
HFRfilter 3 6 5| lowHfilter gbmam.seq.fasta.inf 6 5
filters out the repeats of three successive patterns of length 1 to 3 AND the 6 residual long stretches
containing less than 5 bits of info (thus mostly the leucine-rich areas).
the histogram functionality
The histogram function is designed for pinpointing the odd areas in the query sequence.
There are three kings of histograms meant to be compared: reference histogram (not available in nucleic
acid mode for obvious reasons) gives the a priori expected counts for several lengths on exact matches.
Raw histogram shows the effective count for the observed matches throughout the databank, while the
filtrated histogram give the effective count of reported matches (after scoring and redundancy
suppression).
There is two flavors of histograms, depending whever only the first position in a match is counted or
all the positions within the match. Personaly I prefer the second kind, mostly because the counts decrease
with length.
By pressing the mouse on an histogram in the automat2X11 GUI, the count values (and the residual position and
symbol) are given. It is also possible to change the min and max match length before plotting the histogram.
Areas in the sequence where raw (observed) histogram is very different from the reference (forcasted) histogram
correspond to either repressed or functionnal areas. In the nucleic acids mode, just consider the reference
histogram as flat.
redundancy suppression
The algorithm for detection of redundances involves first the best match score, then the sequence around
it (the ``hash''), then the sequence length, and last the sequence itself. Since the comparison on the sequence
itself is a slow process, it is advised the enable the matrix dithering (it decreases the probability that
different sequences are not sorted appart by the first three criteria).